


Содержание
Перейти к:
 А. М. Якимов,
Е. Е. Тимечко,
А. И. Парамонова,
А. A. Васильева,
Ф. К. Рыбаченко,
А. Д. Рыбаченко,
Д. В. Дмитренко
А. М. Якимов,
Е. Е. Тимечко,
А. И. Парамонова,
А. A. Васильева,
Ф. К. Рыбаченко,
А. Д. Рыбаченко,
Д. В. Дмитренко https://doi.org/10.17749/2077-8333/epi.par.con.2024.210
Перейти к:
В настоящее время проблема эффективной терапии фармакорезистентной эпилепсии остается крайне актуальной. Тяжесть фармакорезистентной эпилепсии, значительные негативные социальные последствия и внезапная смерть при эпилепсии ложатся тяжелым грузом на институт здравоохранения. Хотя за последние десятилетия было разработано множество инновационных противоэпилептических препаратов, единственным эффективным способом лечения фармакорезистентной эпилепсии остается хирургический подход, сопряженный со значительными рисками для здоровья и не гарантирующий при этом свободу от приступов. Камнем преткновения при преодолении данной патологии является недостаток знаний патогенетических механизмов, оставляющий значительную часть больных без качественной медицинской помощи. Различные взгляды на формирование фармакорезистентности при эпилепсии характеризуются комплексностью и наличием пересекающихся молекулярных основ заболевания. В настоящем обзоре проведен анализ существующих концепций о механизмах развития фармакорезистентности при эпилепсии.
Якимов А.М., Тимечко Е.Е., Парамонова А.И., Васильева А.A., Рыбаченко Ф.К., Рыбаченко А.Д., Дмитренко Д.В. Гипотезы развития и стратегии преодоления фармакорезистентности при эпилепсии. Часть I: Гипотезы развития. Эпилепсия и пароксизмальные состояния. 2024;16(4):375-384. https://doi.org/10.17749/2077-8333/epi.par.con.2024.210
Yakimov A.M., Timechko E.E., Paramonova A.I., Vasilieva A.A., Rybachenko F.K., Rybachenko A.D., Dmitrenko D.V. Hypotheses of development and strategies for overcoming drug resistance in epilepsy. Part I: Hypotheses of development. Epilepsy and paroxysmal conditions. 2024;16(4):375-384. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2024.210
Эпилепсия – одно из наиболее распространенных неврологических заболеваний, характеризующееся гетерогенностью и мультифакторной природой, включающее большое количество различных типов приступов и эпилептических синдромов [1][2]. Его распространенность среди мировой популяции оценочно составляет около 1–2% [3]. Основным способом терапии является прием противоэпилептических препаратов (ПЭП), который позволяет добиться длительной ремиссии заболевания [4]. Однако около 30% всех пациентов страдают лекарственно-устойчивой формой эпилепсии, при которой классический вариант терапии малоэффективен [5].
По определению Международной Противоэпилептической Лиги (англ. International League Against Epilepsy, ILAE), фармакорезистентное течение эпилепсии – это такое состояние, при котором испытание двух толерантных и правильно выбранных схем приема ПЭП в виде монотерапии или в комбинации не позволило добиться устойчивого освобождения от приступов [6]. При этом, несмотря на тяжесть заболевания и негативные социальные последствия, механизмы патогенеза эпилепсии в общем и фармакорезистентности в частности на данный момент изучены недостаточно. Более того, фармакорезистентность сама по себе является высоковариабельной и мультифакторной патологией, механизмы которой также дискутабельны [7–9].
Цель – изучение существующих гипотез, которые описывают механизмы развития фармакорезистентности при эпилепсии.
Проведен поиск литературы в базах данных PubMed/MEDLINE, Elsevier, Scopus и Google Scholar за последние 10 лет. Для запросов использовали следующие термины и связанные с ними синонимы: “drug resistant epilepsy”, “refractory epilepsy”, “pharmacoresistant epilepsy” в сочетании с отдельными ключевыми терминами: “mechanisms”, “hypotheses”, “causes’. Также были проанализированы некоторые более ранние работы, отобранные вручную из списков литературы обзорных статей. Все статьи опубликованы на английском и русском языках.
Проанализированы оригинальные исследования, научные и систематические обзоры. В настоящий обзор включены публикации, основной темой которых являлось исследование молекулярно-генетических основ формирования фармакорезистентности при эпилепсии. Исключены дублирующие тексты, а также статьи, к которым отсутствовал полный доступ и не относящиеся к теме исследования. После процедуры отбора (рис. 1) в обзор вошло 76 публикаций.
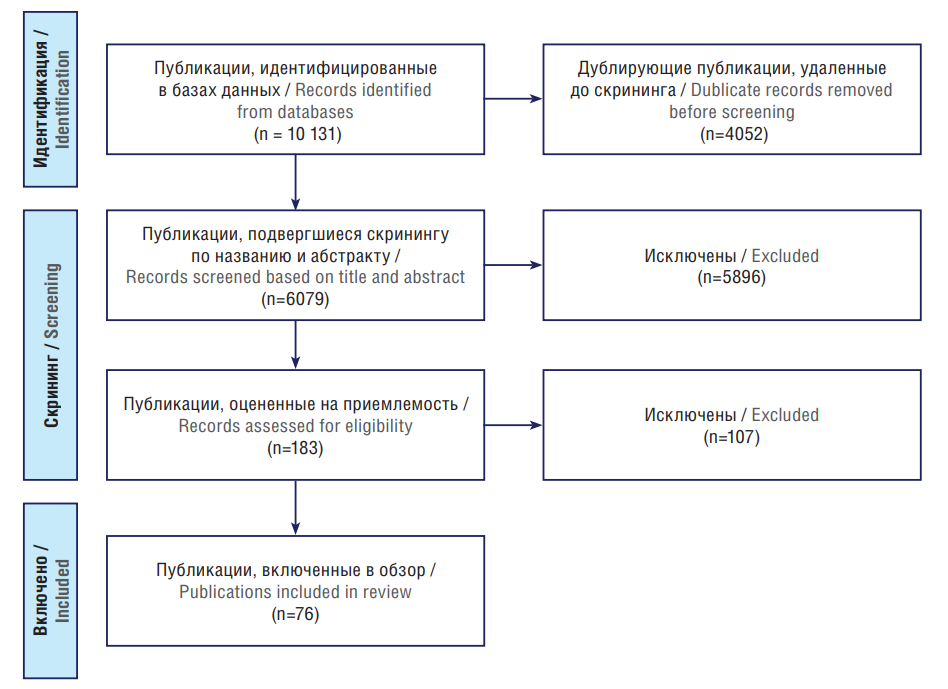
Рисунок 1. Блок-схема PRISMA (англ. Preferred Reporting Items for Systematic reviews and Meta-Analyses) отбора публикаций
Figure 1. PRISMA (Preferred Reporting Items for Systematic reviews and Meta-Analyses) flowchart for selection of publications
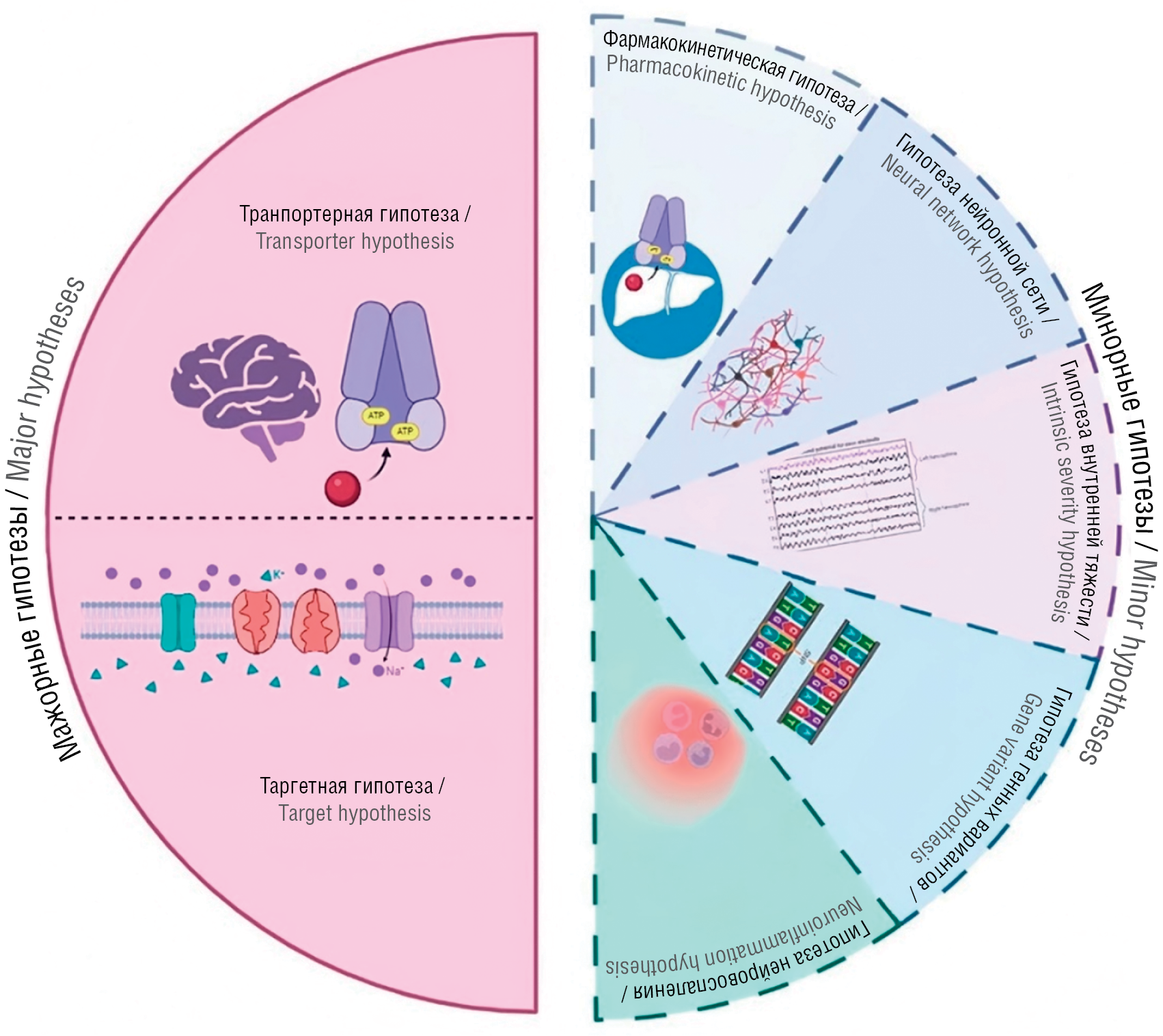
Рисунок 2. Существующие гипотезы фармакорезистентности (рисунок авторов)
Figure 2. Proposed hypotheses for drug resistant epilepsy (drawn by the authors)
На данный момент существует несколько гипотез развития фармакорезистентности при эпилепсии, которые схематически отображены на рисунке 2. Следует отметить, что фармакорезистентность развивается неоднородно в разрезе различных типов эпилепсий. Так, например, пациенты с идиопатической генерализованной эпилепсией легче поддаются медикаментозному лечению – 82% больных достигают ремиссии на фоне терапии. У пациентов с фокальной эпилепсией чаще наблюдается фармакорезистентное течение заболевания – свободы от приступов достигают лишь 11–45% больных после терапии [10][11].
Основными и наиболее цитируемыми являются транспортерная и таргетная гипотезы [12], которые для удобства объединены в блок «мажорные гипотезы». Другие, не так часто обсуждаемые гипотезы сгруппированы в блок «минорные гипотезы».
Транспортерная гипотеза связана с понятием множественной лекарственной устойчивости (МЛУ) – многофакторным явлением, представляющим собой приобретенную устойчивость микроорганизмов и клеток к различным по своей химической природе и механизму действия терапевтическим препаратам [13]. Данное явление обусловлено сверхэкспрессией белков, которые принадлежат к суперсемейству АТФ-связывающих1 транспортных белков (англ. ATP-binding cassette (ABC) transporters) [14]. Известно как минимум 11 ABC-транспортеров, связанных с фенотипом МЛУ, среди которых хорошо известные P-гликопротеины (англ. Р-glycoprotein, P-gp), белки, ассоциированные с МЛУ (англ. multidrug resistance-associated protein, MRP), и белок устойчивости к раку молочной железы (англ. breast cancer resistance protein, BCRP) [15].
Первый описанный механизм, впервые названный МЛУ, был обнаружен у резистентных к химиотерапии раковых клеток. Суть его заключается в сверхэкспрессии эффлюксного транспортера лекарственных препаратов – P-gp, также известного как белок МЛУ [16]. P-gp оказывает значительное влияние на развитие фармакорезистентности при эпилепсии, снижая концентрацию ПЭП в паренхиме мозга [17].
P-gp является трансмембранным транспортерным белком, относящимся к группе белков АТФ-связывающей кассеты. Он представляет собой гликопротеин, состоящий из двух гомологичных половин, которые кодируются геном ABCB1 [18]. Этот белок специфично экспрессируется в барьерных тканях, среди которых гематоэнцефалический барьер (ГЭБ), плацента, кишечник, почки, печень [19]. Помимо этого, P-gp заметно экспрессируется в клетках, обособляющих транспорт препаратов из клеток головного мозга и кровь, а именно: клетки эндотелия, нейроны, астроциты и микроглия [20]. Основной функцией комплекса является обратный транспорт ксенобиотических соединений с широкой субстратной специфичностью. Функционально ABC-транспортеры используют энергию гидролиза АТФ для перемещения множества разнообразных веществ, включая сахара, витамины, гормоны, ксенобиотики, и выполняют тем самым функции поддержания осмотического гомеостаза, захвата питательных веществ, обеспечения устойчивости к токсинам [21].
Хотя до сих пор не до конца ясно, как P-gp распознает свои субстраты, известны условия, которые приводят к снижению концентрации ПЭП в паренхиме мозга [17]:
– ПЭП должен являться субстратом P-gp;
– экспрессия P-gp у пациентов с фармакорезистентной формой эпилепсии должна быть повышена;
– концентрация ПЭП в мозге у больных с фармакорезистентной формой эпилепсии должна быть низкой по сравнению с пациентами с фармакочувствительной формой.
При этом подтвержденными субстратами P-gp являются многие классы терапевтических препаратов [22].
Участие P-gp в явлении фармакорезистентности подтверждается связью его экспрессии с судорожной активностью. Индукция эпилептического статуса повышает экспрессию P-gp и его транспортную активность, приводя к значимому снижению концентрации ПЭП [23][24]. Более того, введение субстратов P-gp, таких как глутамат, вызывает нейровоспаление, значительно увеличивая экспрессию транспортера [25]. Способны модулировать экспрессию P-gp также и ПЭП, а именно фенобарбитал, фенитоин и карбамазепин [19][26][27]. Прием же таких препаратов, как леветирацетам, топирамат и фенитоин, усиливал функцию P-gp, что снижало общий терапевтический эффект, при этом вальпроевая кислота не проявляла взаимодействия с P-gp [28].
В целом при ряде неврологических заболеваний, для которых характерно снижение эффективности медикаментозной терапии, наблюдается повышение экспрессии P-gp [29]. Причем увеличенные уровни экспрессии и активности P-gp могут быть связаны с разнообразными сигнальными путями, среди которых фактор некроза опухоли альфа (англ. tumor necrosis factor alpha, TNF-α), протеинкиназа С бета (англ. protein kinase C beta, PKC-β), сфингозин-1-фосфат (англ. sphingosine-1-phosphate, S1P), фактор роста эндотелия сосудов (англ. vascular endothelial growth factor, VEGF), протоонкоген тирозин-протеинкиназа Src, транскрипционный ядерный фактор каппа В (англ. nuclear factor kappa B, NF-κB), рецептор прегнана X (англ. pregnane X receptor, PXR), химерный антигенный рецептор (англ. chimeric antigen receptor, CAR) [27][30]. Аберрантная активность этих сигнальных путей также может быть вызвана различными факторами, включая гипоксию, индуцирующую появление провоспалительных медиаторов и активных форм кислорода [31], повышенное содержание глутамата [27].
Суть таргетной гипотезы заключается в структурных изменениях клеточных мишеней ПЭП, что вызывает снижение чувствительности к терапии [32]. В зависимости от типа воздействия лекарственных препаратов выделяют следующие группы [33]:
– модуляторы потенциал-зависимых ионных каналов;
– потенциирование ГАМКергической2 трансмиссии;
– множественный механизм действия;
– другой механизм действия.
Непосредственно для эпилепсии характерны изменения в различных каналах [34]. Ниже приведены примеры таких патологий.
Натриевые каналы
На данный момент потенциал-зависимые натриевые каналы являются наиболее распространенной и часто используемой мишенью ПЭП [35]. Они представлены гетеромерными трансмембранными белками, активирующим событием для которых служит деполяризация мембраны. Функционально данные каналы ответственны за генерацию и распространение потенциалов действия в нейронах и иных электровозбудимых клетках. Сам канал образован одной основной α-субъединицей, которая связана с одной или несколькими из четырех вспомогательных β-субъединиц [36]. Известно, что носительство патогенных вариантов в генах SCN1A, SCN1B, SCN2A, SCN3A, SCN8A и SCN9A ассоциировано со значительной частью всех случаев развития фармакорезистентной формы эпилепсии в детстве и младенчестве [37].
В качестве примеров можно привести ряд однонуклеотидных вариантов (ОНВ) в генах, кодирующих α-субъединицы натриевых каналов. Так, ОНВ rs3812718 гена SCN1A ответственен за снижение чувствительности к карбамазепину, а носительство полиморфного варианта rs2304016 гена SCN2A коррелирует с фармакорезистентностью [38][39]. Интересна мутация C121W в канале гена SCN1B, которая ассоциирована с развитием генерализованной эпилепсии с фебрильными приступами плюс (англ. genetic epilepsy with febrile seizures plus, GEFS+), а также снижает чувствительность канала к фенитоину, причем такое изменение не связано напрямую с сайтом связывания лекарства [40].
В целом развитие фармакорезистентности, связанной с каналопатиями, не ограничивается точечными изменениями в генах. Так, одной из причин снижения чувствительности к ПЭП, основанного на блокировке натриевых каналов, может служить измененное соотношение экспрессии субъединиц этих каналов. В норме соотношение экспрессии различных подтипов субъединиц характерно для определенных онтогенетических, региональных и субклеточных паттернов [41]. При наличии заболевания эти паттерны изменяются по следующим характеристикам [42]:
– изменения экспрессии локализованы в определенных областях;
– изменяется экспрессия мРНК;
– наблюдаются разные кинетика и специфика для каждого подтипа.
Иллюстрацией такого паттерн-зависимого ингибирования может служить работа, проделанная на пилокарпиновой хронической модели эпилепсии крыс, которая показала, что нейроны зубчатой извилины гиппокампа больше подвержены потере чувствительности к ПЭП, чем нейроны региона CA1 [43]. О величине изменения экспрессии отдельных субъединиц друг относительно друга можно судить по изменению отношения субъединиц Nav1.1 (SCN1A) и Nav1.2 (SNC2A) в различных областях мозга, где соотношение 1:2 наблюдалось в контрольных образцах и могло изменяться вплоть до 1:3 в соответствующих участках с эпилептогенной активностью [44].
При этом в отдельных исследованиях показан неравномерный эффект ПЭП, действующих на натриевые каналы, при фармакорезистентности. Так, если способности фенитоина и ламотриджина купировать приступы при фармакорезистентной эпилепсии были значительно снижены, то эффекты вальпроевой кислоты не ингибировались [45]. Ингибирования эффектов вальпроевой кислоты не наблюдалось и при сравнении пациентов со склерозом гиппокампа и без него, а также в тканях региона CA1 гиппокампа и неокортекса [46]. Схожие данные получены в исследовании, где эффективность карбамазепина понижалась в случае фармакорезистентности, а эффективность вальпроатов оставалась неизменной [47].
Калиевые каналы
Потенциал-зависимые калиевые каналы контролируют мембранный потенциал покоя и обеспечивают реполяризацию потенциала действия, создавая токи ионов калия наружу и ограничивая возбудимость нейронов [48]. За последние несколько лет непосредственно с эпилепсией было ассоциировано как минимум 19 мутаций в генах, кодирующих калиевые каналы, которые относятся к различным функциональным и структурным группам. В целом с патогенезом эпилепсии связывают следующие гены, кодирующие калиевые каналы: KCNA1, KCNA2, KCNB1, KCNC1, KCNH1, KCNQ2, KCNQ3, KCNT1, KCNT2, KCNT1 и KCNMA1 [49].
Такое высокое число изменений в калиевых каналах, ассоциированных с эпилепсией, объясняется крайне высоким молекулярным и функциональным разнообразием. Однако повышение восприимчивости к приступам связано не только с врожденными мутациями, но и с модификациями свойств каналов. Вследствие широкого разнообразия измененных каналов, ассоциированных с эпилепсией, для каждого из них описан отдельный фенотип, связанный с потерей функции [50]. Однако существует и ситуация, названная «неожиданным парадоксом»: она вызвана мутацией в гене KCNT1 и приводит к усилению функции калиевого канала, что является причиной возникновения эпилепсии [51].
Кальциевые каналы
Кальциевые каналы – ионные поры, обладающие избирательной проницаемостью для ионов кальция. Они представляют собой гомо- или гетерополимерные мембранные комплексы, в основе каждого из которых лежит субъединица α1, определяющая тип канала. Всего идентифицировано 10 различных субъединиц α1, которые могут быть объединены в комплекс с субъединицами β, α2δ и γ [52].
В нервной системе существует большое разнообразие кальциевых каналов, направленных на выполнение различных специфичных функций. Оно обусловлено альтернативным сплайсингом и множеством комбинаций объединения субъединиц [53]. Кальциевые каналы обес- печивают различные процессы нейрональной активности, представляя собой один из основных компонентов возбудимости нейронов [54].
Одним из основных процессов нейродегенерации является нарушение гомеостаза внутриклеточного кальция, например в случае чрезмерного высвобождения глутамата [55]. Повышенное содержание внутриклеточного кальция наблюдается также и при посттравматической эпилепсии на фоне дисфункции и гибели нейронов [56]. Результатом таких нарушений становятся повышенная восприимчивость нейронов к приступам и потенциальное развитие фармакорезистентности [57]. Также мутации в генах, кодирующих различные субъединицы кальциевых каналов, ассоциированы с появлением нейродегенеративных заболеваний, образуя эпилептогенные эффекты без внятного единого механизма, объясняющего их [58].
Глутамат- и ГАМКергические системы
ГАМК- и глутаматергические системы являются основными тормозящими и возбуждающими системами соответственно [59]. Нарушение баланса возбуждающей и тормозящей систем часто встречается при неврологических заболеваниях, в частности при эпилепсии, опосре- дуя процессы иктогенеза и эпилептогенеза [60].
Эпилептический статус приводит к реорганизации нейронной сети и физиологическим перестройкам в мозге [61]. Эти перестройки включают развивающийся дисбаланс возбуждающих и тормозящих нейромедиаторов, который подтверждается как на уровне иммуногистохимических исследований [62], так и в изменениях экспрессии генов, кодирующих рецепторы ГАМК и глутамата [63]. Помимо этого, изменяется и экспрессия генов, регулирующих их высвобождение и метаболизацию [64].
Последствиями такого дисбаланса являются дисфункция синаптической передачи, нарушение когнитивных и двигательных функций и повреждение нейронов [59]. Метаболические пути этих двух нейромедиаторов неразрывно связаны, т.к. глутамат является предшественником ГАМК. Почти вся поступающая в мозг глюкоза служит субстратом для превращения в глутамат [65]. При этом нарушение цикла «глутамат – глутамин – ГАМК» наблюдается у пациентов с рефрактерной формой эпилепсии [66]. Также причиной развития фармакорезистентности становятся различные мутации в генах, участвующих в метаболизме ГАМК и глутамата [67].
Так, в срезах эпилептогенной и перифокальной зоны от пациентов с посттравматической фармакорезистентной эпилепсией наблюдался дисбаланс глутамат- и ГАМК-ергических систем. Выявлено повышенное содержание GAD65 (фермент, превращающий глутамат в ГАМК) и Vglut2 (везикулярный транспортер глутамата) в эпилептическом очаге, а также в сером веществе височной коры. Повышение экспрессии GAD65 свидетельствует об активации компенсаторных механизмов при гибели ГАМКергических нейронов. Кроме того, повышение экспрессии Vglut2 наблюдалось в перифокальной зоне. Нарушалось также соотношение про- и противоапоптических ферментов: в эпилептическом очаге, сером и белом веществах височной доли снижалось содержание противоапоптического белка Bcl-2, но повышалось содержание проапоптического белка CASP8 в очаге и перифокальной зоне [68].
Мутации в гене GAT1 – ГАМК-переносчике, отвечающем за обратный захват нейромедиатора из синапса, приводят к снижению транспортной активности и обратного захвата ГАМК из синапсов, что вносит вклад в развитие приступов при эпилепсии [69][70]. Также мутации могут встречаться и в генах субъединиц самого ГАМК-рецептора, каждая из которых может принимать участие в различных формах эпилепсии [71]. Хотя эти мутации и вызывают дефекты, связанные с гиперполяризацией, опосредованной ГАМК, сам механизм нарушения функционирования значительно сложнее и включает процессы деградации эндоплазматического ретикулума, нонсенс-опосредованной деградации мРНК, нарушения внутриклеточного транспорта, а также стресс эндоплазматического ретикулума [72]. Некоторые же мутации, например в генах GABRG2, GABRA1 и GABRB3, могут быть ответственны за развитие фенотипа, схожего с синдромом Драве [73].
При эпилепсиях, в т.ч. рефрактерных формах, наблюдаются нарушения клиренса глутамата – например, вследствие снижения экспрессии GLAST и GLT1 [74], приводящего к его накоплению в синаптической щели. Такой патологический процесс вызывает постоянный возбуждающий глутаматергический поток в нейронах, приводя к гиперактивности, влекущей за собой развитие эпилептических приступов, а также эксайтотоксичность [75].
Фармакокинетическая гипотеза постулирует, что за явление фармакорезистентности ответственна сверхэкспрессия эффлюксных транспортеров в периферических органах, таких как кишечник, печень, почки, которые снижают уровень ПЭП в плазме крови, сокращая количество препарата, доступного для преодоления ГЭБ и достижения головного мозга. Такое предположение подкрепляется повышенным клиренсом у пациентов с фармакорезистентной эпилепсией радиофармпрепарата 99mTc-гексакис-2-метоксиизобутилизонитрила – субстрата P-gp [76]. В другом случае авторы наблюдали устойчивые субтерапевтические уровни ПЭП в плазме крови, что совпадало со сверхэкспрессией P-gp в эндотелии, астроцитах и нейронах резецированной ткани [77].
Таким образом, устойчивые низкие уровни ПЭП не могут быть объяснены только сверхэкспрессией в ткани мозга или ГЭБ. Это позволяет предположить наличие сверхэкспрессии P-gp также в периферических органах [76].
Некоторое подтверждение данной гипотезы можно найти в исследованиях, показывающих стойкие низкие уровни ПЭП вне зависимости от экспрессии P-gp. Так, выявлено снижение средней концентрации свободного ПЭП в плазме крови [78].
Однако такие же данные не удается продемонстрировать на животных моделях [79]. Кроме того, в других исследованиях не показано непосредственной связи между повышением уровня экспрессии P-gp в периферических органах и низкими концентрациями в плазме крови при фармакорезистентной эпилепсии [80].
Косвенно опровергает указанную гипотезу и тот факт, что и в группе пациентов, чувствительных к терапии, и в группе резистентных больных побочные эффекты от приема ПЭП проявляются в одинаковой степени [81][82].
Гипотеза нейронной сети основана на структурных изменениях под действием генов и микроокружения, включающих разрастание аксонов, синаптическую реорганизацию, нейрогенез и глиоз, вызванные эпилептической активностью головного мозга. Такие изменения формируют аномалии нейронной сети, проявляющиеся в подавлении эндогенной противоэпилептической системы и попадания ПЭП к их целям [83].
Основные доказательства гипотезы получены на исследованиях гиппокампального склероза, который часто наблюдается у пациентов с фармакорезистентным течением заболевания и, по всей видимости, участвует в механизмах резистентности [84]. Имеются и данные о том, что функциональные изменения пирамидальных нейронов гиппокампа и зубчатой извилины, развивающиеся в ответ на потерю нейронов, играют роль в формировании резистентности к ПЭП [85][86]. Также известно, что при резекции пораженной части височной доли и дальнейшем проведении медикаментозного лечения приступы удается купировать [87].
Однако недостатком данной гипотезы является то, что структурные изменения в головном мозге приводят к рефрактерности не у всех пациентов [12].
Еще одной теорией формирования фармакорезистентной формы эпилепсии является гипотеза внутренней тяжести. Она утверждает, что нейробиологические факторы, обусловливающие повышенную тяжесть заболевания, приводят к резистентности к лекарственным препаратам [9]. Основой гипотезы служит наблюдение, в котором у пациентов с впервые диагностированной эпилепсией с более высокой частотой приступов до начала лечения вероятность ремиссии была ниже [88]. Под термином «тяжесть» в данном случае понимается влияние заболевания на биологические, физические или психосоциальные функции, определяемое частотой эпилептических приступов [89][90].
Однако у гипотезы есть слабые места, связанные с тем, что если сначала только у 7% пациентов, впервые получивших лечение, сразу наблюдается резистентность к лекарствам, то в течение эпилептогенеза она развивается еще у 15–20% [91]. Каждому пятому пациенту после изменения схемы медикаментозного лечения удается освободиться от эпилептических приступов. Таким образом, резистентность может быть обратимой, а внутренняя тяжесть эпилепсии может со временем снижаться [92].
Помимо этого, существует ряд ограничений данной гипотезы [92]:
– отсутствуют исследования нейробиологических основ;
– некоторым пациентам с более высокой частотой приступов в начале лечения требуется более высокая концентрация ПЭП в сыворотке крови (предположение о том, что гипотеза не способна объяснить или предсказать резистентность);
– данные магнитно-резонансной томографии, полученные в группе детей с височной эпилепсией, спустя 7 и 14 лет показали поражения мозга, а частота приступов и ранняя ремиссия не смогли спрогнозировать исход приступов.
Гипотеза генных вариантов является, скорее, закономерным дополнением таргетной гипотезы и утверждает, что вариации генов, связанные с фармакокинетикой и фармакодинамикой ПЭП, обусловливают возникающую фармакорезистентность [93].
Некоторые примеры, связанные с метаболизмом ПЭП и демонстрирующие ассоциацию носительства аллельного варианта гена CYP2C9 со снижением эффективной дозы фенитоина, описаны в посвященной фармакорезистентной эпилепсии обзорной статье [12].
Гипотеза основана на установленных фактах увеличения проницаемости ГЭБ, что показано на эпилептических очагах, индуцированных посредством его искусственной дисфункции [94]. Более того, опосредованная нейровоспалением дисфункция ГЭБ усиливает и экспрессию P-gp.
В целом предполагается три варианта развития резистентности к лекарствам за счет нейровоспаления [8]:
– прямое воздействие на эндотелий сосудов, выражающееся в разрушении плотных контактов между клетками эндотелия, индукции аномального ангиогенеза и окислительном стрессе;
– стимуляция экспрессии P-gp в эндотелиальных клетках за счет действия провоспалительных медиаторов;
– посттрансляционная модификация потенциал-зависимых ионных каналов провоспалительными медиаторами, что снижает чувствительность рецепторов к субстрату.
Вместе с тем имеются и данные, наоборот, говорящие о снижении уровней биомаркеров нейровоспаления у пациентов с фармакорезистентной формой эпилепсии [95].
Фармакорезистентность при эпилепсии представляет спектр различных патологических состояний. Из этого вытекает несколько гипотез, объясняющих механизмы ее развития. Однако ни одна из существующих в настоящее время теорий не позволяет в точности описать процессы, приводящие к фармакорезистентности. Хотя эти гипотезы самодостаточны относительно друг друга, можно проследить наличие взаимосвязей между ними на различных уровнях организации. При этом благодаря текущим исследованиям представляется возможным преодолевать рефрактерность к лекарственным препаратам тем или иным способом, что говорит о необходимости продолжения исследований в данном направлении.
1. АТФ – аденозинтрифосфат.
2. ГАМК – гамма-аминомасляная кислота.
1. Schmidt D., Schachter S.C. Drug treatment of epilepsy in adults. BMJ. 2014; 348: g254. https://doi.org/10.1136/BMJ.G254.
2. Насырова Р.Ф., Сивакова Н.А., Липатова Л.В. и др. Биологические маркеры эффективности и безопасности противоэпилептических препаратов: фармакогенетика и фармакокинетика. Сибирское медицинское обозрение. 2017; 1: 17–25. https://doi.org/10.20333/2500136-2017-1-17-25.
3. Falco-Walter J. Epilepsy – definition, classification, pathophysiology, and epidemiology. Semin Neurol. 2020; 40 (6): 617–23. https://doi.org/10.1055/S-0040-1718719.
4. Johannessen Landmark C., Johannessen S.I., Patsalos P.N. Therapeutic drug monitoring of antiepileptic drugs: current status and future prospects. Expert Opin Drug Metab Toxicol. 2020; 16 (3): 227–38. https://doi.org/10.1080/17425255.2020.1724956.
5. Mesraoua B., Brigo F., Lattanzi S., et al. Drug-resistant epilepsy: definition, pathophysiology, and management. J Neurol Sci. 2023; 452: 120766. https://doi.org/10.1016/j.jns.2023.120766.
6. Kwan P., Arzimanoglou A., Berg A.T., et al. Definition of drug resistant epilepsy: Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia. 2010; 51 (6): 1069–77. https://doi.org/10.1111/j.1528-1167.2009.02397.x.
7. Lerche H. Drug-resistant epilepsy – time to target mechanisms. Nat Rev Neurol. 2020; 16 (11): 595–6. https://doi.org/10.1038/s41582-020-00419-y.
8. Bazhanova E.D., Kozlov A.A., Litovchenko A.V. Mechanisms of drug resistance in the pathogenesis of epilepsy: role of neuroinflammation. A literature review. Brain Sci. 2021; 11 (5): 663. https://doi.org/10.3390/brainsci11050663.
9. Łukawski K., Czuczwar S.J. Understanding mechanisms of drug resistance in epilepsy and strategies for overcoming it. Expert Opin Drug Metab Toxicol. 2021; 17 (9): 1075–90. https://doi.org/10.1080/17425255.2021.1959912.
10. Sampaio L.P. Ketogenic diet for epilepsy treatment. Arq Neuropsiquiatr. 2016; 74 (10): 842–8. https://doi.org/10.1590/0004-282X20160116.
11. Bartolini E., Ferrari A.R., Lattanzi S., et al. Drug-resistant epilepsy at the age extremes: disentangling the underlying etiology. Epilepsy Behav. 2022; 132: 108739. https://doi.org/10.1016/j.yebeh.2022.108739.
12. Tang F., Hartz A.M.S., Bauer B. Drug-resistant epilepsy: multiple hypotheses, few answers. Front Neurol. 2017; 8: 301. https://doi.org/10.3389/fneur.2017.00301.
13. Catalano A., Iacopetta D., Ceramella J., et al. Multidrug resistance (MDR): a widespread phenomenon in pharmacological therapies. Molecules. 2022; 27 (3): 616. https://doi.org/10.3390/molecules27030616.
14. Pérez-Pérez D., Frías-Soria C.L., Rocha L. Drug-resistant epilepsy: From multiple hypotheses to an integral explanation using preclinical resources. Epilepsy Behav. 2021; 121 (Pt B): 106430. https://doi.org/10.1016/j.yebeh.2019.07.031.
15. Liu X. ABC family transporters. Adv Exp Med Biol. 2019; 1141: 13–100. https://doi.org/10.1007/978-981-13-7647-4_2.
16. Brandt C., Bethmann K., Gastens A.M., Löscher W. The multidrug transporter hypothesis of drug resistance in epilepsy: proof-of-principle in a rat model of temporal lobe epilepsy. Neurobiol Dis. 2006; 24 (1): 202–11. https://doi.org/10.1016/j.nbd.2006.06.014.
17. Garg N., Joshi R., Medhi B. A novel approach of targeting refractory epilepsy: need of an hour. Brain Res Bull. 2020; 163: 14–20. https://doi.org/10.1016/j.brainresbull.2020.07.012.
18. Kurre D., Dang P.X., Le L.T.M., et al. Structural insight into binding site access and ligand recognition by human ABCB1. BioRxiv. 2024; Aug 12: 2024.08.12.607598. https://doi.org/10.1101/2024.08.12.607598.
19. Elmeliegy M., Vourvahis M., Guo C., Wang D.D. Effect of P-glycoprotein (P-gp) inducers on exposure of P-gp substrates: review of clinical drug–drug interaction studies. Clin Pharmacokinet. 2020; 59 (6): 699–714. https://doi.org/10.1007/s40262-020-00867-1.
20. Fonseca-Barriendos D., Pérez-Pérez D., Fuentes-Mejía M., et al. Protein expression of P-glycoprotein in neocortex from patients with frontal lobe epilepsy. Epilepsy Res. 2022; 181: 106892. https://doi.org/10.1016/j.eplepsyres.2022.106892.
21. Mossel P., Arif W.M., De Souza G.S., et al. Quantification of P-glycoprotein function at the human blood-brain barrier using [18F] MC225 and PET. Eur J Nucl Med Mol Imaging. 2023; 50 (13): 3917–27. https://doi.org/10.1007/S00259-023-06363-5.
22. Dong J., Qin Z., Zhang W.D., et al. Medicinal chemistry strategies to discover P-glycoprotein inhibitors: an update. Drug Resist Updat. 2020; 49: 100681. https://doi.org/10.1016/j.drup.2020.100681.
23. Kaur M., Gupta T., Gupta M., et al. Expressional study of permeability glycoprotein and multidrug resistance protein 1 in drug-resistant mesial temporal lobe epilepsy. Basic Clin Neurosci. 2023; 14 (5): 615–30. https://doi.org/10.32598/bcn.2021.2554.3.
24. Langeh U., Chawla P., Gupta G.D., Singh S. A novel approach to refractory epilepsy by targeting pgp peripherally and centrally: therapeutic targets and future perspectives. CNS Neurol Disord Drug Targets. 2020; 19 (10): 741–9. https://doi.org/10.2174/1871527319999200819093109.
25. Enrique A.V., Di Ianni M.E., Goicoechea S., et al. New anticonvulsant candidates prevent P-glycoprotein (P-gp) overexpression in a pharmacoresistant seizure model in mice. Epilepsy Behav. 2021; 121 (Pt B): 106451. https://doi.org/10.1016/j.yebeh.2019.106451.
26. Wen T., Liu Y.C., Yang H.W., et al. Effect of 21-day exposure of phenobarbital, carbamazepine and phenytoin on P-glycoprotein expression and activity in the rat brain. J Neurol Sci. 2008; 270 (1–2): 99–106. https://doi.org/10.1016/j.jns.2008.02.016.
27. Ke X.J., Cheng Y.F., Yu N., Di Q. Effects of carbamazepine on the P-gp and CYP3A expression correlated with PXR or NF-κB activity in the bEnd.3 cells. Neurosci Lett. 2019; 690: 48–55. https://doi.org/10.1016/j.neulet.2018.10.016.
28. Moerman L., Wyffels L., Slaets D., et al. Antiepileptic drugs modulate P-glycoproteins in the brain: a mice study with (11) C-desmethylloperamide. Epilepsy Res. 2011; 94 (1–2): 18–25. https://doi.org/10.1016/j.eplepsyres.2010.12.013.
29. Huang L., Li B., Li X., et al. Significance and mechanisms of P-glycoprotein in central nervous system diseases. Curr Drug Targets. 2019; 20 (11): 1141–55. https://doi.org/10.2174/1389450120666190308144448.
30. Ding Y., Wang R., Zhang J., et al. Potential regulation mechanisms of P-gp in the blood-brain barrier in hypoxia. Curr Pharm Des. 2019; 25 (10): 1041–51. https://doi.org/10.2174/1381612825666190610140153.
31. Mohamed L.A., Markandaiah S.S., Bonanno S., et al. Excess glutamate secreted from astrocytes drives upregulation of P-glycoprotein in endothelial cells in amyotrophic lateral sclerosis. Exp Neurol. 2019; 316: 27–38. https://doi.org/10.1016/j.expneurol.2019.04.002.
32. Fonseca-Barriendos D., Frías-Soria C.L., Pérez-Pérez D., et al. Drug-resistant epilepsy: drug target hypothesis and beyond the receptors. Epilepsia Open. 2022; 7 (Suppl. 1): S23–33. https://doi.org/10.1002/epi4.12539.
33. Kim H., Kim D.W., Lee S.T., et al. Antiepileptic drug selection according to seizure type in adult patients with epilepsy. J Clin Neurol. 2020; 16 (4): 547–55. https://doi.org/10.3988/jcn.2020.16.4.547.
34. Bartolini E., Campostrini R., Kiferle L., et al. Epilepsy and brain channelopathies from infancy to adulthood. Neurol Sci. 2020; 41 (4): 749–61. https://doi.org/10.1007/s10072-019-04190-x.
35. Kobayashi K., Endoh F., Ohmori I., Akiyama T. Action of antiepileptic drugs on neurons. Brain Dev. 2020; 42 (1): 2–5. https://doi.org/10.1016/j.braindev.2019.07.006.
36. Catterall W.A. Voltage gated sodium and calcium channels: discovery, structure, function, and pharmacology. Channels. 2023; 17 (1): 2281714. https://doi.org/10.1080/19336950.2023.2281714.
37. Parrini E., Marini C., Mei D., et al. Diagnostic targeted resequencing in 349 patients with drug-resistant pediatric epilepsies identifies causative mutations in 30 different genes. Hum Mutat. 2017; 38 (2): 216–25. https://doi.org/10.1002/humu.23149.
38. Zhao G.X., Zhang Z., Cai W.K., et al. Associations between CYP3A4, CYP3A5 and SCN1A polymorphisms and carbamazepine metabolism in epilepsy: a meta-analysis. Epilepsy Res. 2021; 173: 106615. https://doi.org/10.1016/j.eplepsyres.2021.106615.
39. Lin C.H., Ho C.J., Lu Y.T., Tsai M.H. Response to sodium channel blocking antiseizure medications and coding polymorphisms of sodium channel genes in Taiwanese epilepsy patients. BMC Neurol. 2021; 21 (1): 367. https://doi.org/10.1186/s12883-021-02395-2.
40. Al-Ward H., Liu C.Y., Liu N., et al. Voltage-gated sodium channel β1 gene: an overview. Hum Hered. 2020; 85 (3–6): 101–9. https://doi.org/10.1159/000516388.
41. Barbieri R., Nizzari M., Zanardi I., et al. Voltage-gated sodium channel dysfunctions in neurological disorders. Life. 2023; 13 (5): 1191. https://doi.org/10.3390/life13051191.
42. Bartolomei F., Gastaldi M., Massacrier A., et al. Changes in the mRNAs encoding subtypes I, II and III sodium channel alpha subunits following kainate-induced seizures in rat brain. J Neurocytol. 1997; 26 (10): 667–78. https://doi.org/10.1023/a:1018549928277.
43. Schaub C., Uebachs M., Beck H. Diminished response of CA1 neurons to antiepileptic drugs in chronic epilepsy. Epilepsia. 2007; 48 (7): 1339–50. https://doi.org/10.1111/j.1528-1167.2007.01103.x.
44. Lombardo A.J., Kuzniecky R., Powers R.E., Brown G.B. Altered brain sodium channel transcript levels in human epilepsy. Brain Res Mol Brain Res. 1996; 35 (1–2): 84–90. https://doi.org/10.1016/0169-328x(95)00194-w.
45. Remy S., Urban B.W., Elger C.E., Beck H. Anticonvulsant pharmacology of voltage-gated Na+ channels in hippocampal neurons of control and chronically epileptic rats. Eur J Neurosci. 2003; 17 (12): 2648–58. https://doi.org/10.1046/j.1460-9568.2003.02710.x.
46. Vreugdenhil M., Van Veelen C.W.M., Van Rijen P.C., et al. Effect of valproic acid on sodium currents in cortical neurons from patients with pharmaco-resistant temporal lobe epilepsy. Epilepsy Res. 1998; 32 (1–2): 309–20. https://doi.org/10.1016/s0920-1211(98)00061-8.
47. Vreugdenhil M., Wadman W.J. Modulation of sodium currents in rat CA1 neurons by carbamazepine and valproate after kindling epileptogenesis. Epilepsia. 1999; 40 (11): 1512–22. https://doi.org/10.1111/j.1528-1157.1999.tb02034.x.
48. de Lera Ruiz M., Kraus R.L. Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J Med Chem. 2015; 58 (18): 7093–118. https://doi.org/10.1021/jm501981g.
49. Zhao T., Wang L., Chen F. Potassium channel-related epilepsy: pathogenesis and clinical features. Epilepsia Open. 2024; 9 (3): 891–5. https://doi.org/10.1002/epi4.12934.
50. Gao K., Lin Z., Wen S., Jiang Y. Potassium channels and epilepsy. Acta Neurol Scand. 2022; 146 (6): 699–707. https://doi.org/10.1111/ane.13695.
51. Niday Z., Tzingounis A.V. Potassium channel gain of function in epilepsy: an unresolved paradox. Neuroscientist. 2018; 24 (4): 368–80. https://doi.org/10.1177/1073858418763752.
52. Catterall W.A., Lenaeus M.J., Gamal El-Din T.M. Structure and pharmacology of voltage-gated sodium and calcium channels. Annu Rev Pharmacol Toxicol. 2020; 60: 133–54. https://doi.org/10.1146/annurev-pharmtox-010818-021757.
53. Lipscombe D., Andrade A., Allen S.E. Alternative splicing: functional diversity among voltage-gated calcium channels and behavioral consequences. Biochim Biophys Acta. 2013; 1828 (7): 1522–9. https://doi.org/10.1016/j.bbamem.2012.09.018.
54. Kessi M., Chen B., Peng J., et al. Calcium channelopathies and intellectual disability: a systematic review. Orphanet J Rare Dis. 2021; 16 (1): 219. https://doi.org/10.1186/S13023-021-01850-0.
55. Verma M., Lizama B.N., Chu C.T. Excitotoxicity, calcium and mitochondria: a triad in synaptic neurodegeneration. Transl Neurodegener. 2022; 11 (1): 3. https://doi.org/10.1186/S40035-021-00278-7.
56. Gurkoff G.G., Shahlaie K., Lyeth B.G. In vitro mechanical strain trauma alters neuronal calcium responses: implications for posttraumatic epilepsy. Epilepsia. 2012; 53 (Suppl. 1): 53–60. https://doi.org/10.1111/j.1528-1167.2012.03475.x.
57. Steinlein O.K. Calcium signaling and epilepsy. Cell Tissue Res. 2014; 357 (2): 385–93. https://doi.org/10.1007/s00441-014-1849-1.
58. Gambardella A., Labate A. The role of calcium channel mutations in human epilepsy. Prog Brain Res. 2014; 213: 87–96. https://doi.org/10.1016/B978-0-444-63326-2.00004-1.
59. Sears S.M.S., Hewett S.J. Influence of glutamate and GABA transport on brain excitatory/inhibitory balance. Exp Biol Med. 2021; 246 (9): 1069–83. https://doi.org/10.1177/1535370221989263.
60. Sarlo G.L., Holton K.F. Brain concentrations of glutamate and GABA in human epilepsy: a review. Seizure. 2021; 91: 213–27. https://doi.org/10.1016/j.seizure.2021.06.028.
61. Trinka E., Leitinger M. Management of status epilepticus, refractory status epilepticus, and super-refractory status epilepticus. Continuum. 2022; 28 (2): 559–602. https://doi.org/10.1212/con.0000000000001103.
62. Mathern G.W., Pretorius J.K., Leite J.P., et al. Hippocampal AMPA and NMDA mRNA levels and subunit immunoreactivity in human temporal lobe epilepsy patients and a rodent model of chronic mesial limbic epilepsy. Epilepsy Res. 1998; 32 (1–2): 154–71. https://doi.org/10.1016/S0920-1211(98)00048-5.
63. Crino P.B., Duhaime A.C., Baltuch G., White R. Differential expression of glutamate and GABA-A receptor subunit mRNA in cortical dysplasia. Neurology. 2001; 56 (7): 906–13. https://doi.org/10.1212/wnl.56.7.906.
64. Adams C.E., Yonchek J.C., Schulz K.M., et al. Reduced Chrna7 expression in mice is associated with decreases in hippocampal markers of inhibitory function: implications for neuropsychiatric diseases. Neuroscience. 2012; 207: 274–82. https://doi.org/10.1016/j.neuroscience.2012.01.033.
65. Andersen J.V., Schousboe A., Verkhratsky A. Astrocyte energy and neurotransmitter metabolism in Alzheimer’s disease: Integration of the glutamate/GABA-glutamine cycle. Prog Neurobiol. 2022; 217: 102331. https://doi.org/10.1016/j.pneurobio.2022.102331.
66. Çavuş I., Romanyshyn J.C., Kennard J.T., et al. Elevated basal glutamate and unchanged glutamine and GABA in refractory epilepsy: microdialysis study of 79 patients at the yale epilepsy surgery program. Ann Neurol. 2016; 80 (1): 35–45. https://doi.org/10.1002/ana.24673.
67. Feng Y., Wei Z.H., Liu C., et al. Genetic variations in GABA metabolism and epilepsy. Seizure. 2022; 101: 22–9. https://doi.org/10.1016/j.seizure.2022.07.007.
68. Sazhina T.A., Sitovskaya D.A., Zabrodskaya Y.M., Bazhanova E.D. Functional imbalance of glutamate- and GABAergic neuronal systems in the pathogenesis of focal drug-resistant epilepsy in humans. Bull Exp Biol Med. 2020; 168 (4): 529–32. https://doi.org/10.1007/s10517-020-04747-3.
69. Carvill G.L., McMahon J.M., Schneider A., et al. Mutations in the GABA transporter SLC6A1 cause epilepsy with myoclonic-atonic seizures. Am J Hum Genet. 2015; 96 (5): 808–15. https://doi.org/10.1016/j.ajhg.2015.02.016.
70. Mattison K.A., Butler K.M., Inglis G.A.S., et al. SLC6A1 variants identified in epilepsy patients reduce γ-aminobutyric acid transport. Epilepsia. 2018; 59 (9): e135–41. https://doi.org/10.1111/epi.14531.
71. Kang J.Q. Defects at the crossroads of GABAergic signaling in generalized genetic epilepsies. Epilepsy Res. 2017; 137: 9–18. https://doi.org/10.1016/j.eplepsyres.2017.08.013.
72. Hirose S. Mutant GABA(A) receptor subunits in genetic (idiopathic) epilepsy. Prog Brain Res. 2014; 213: 55–85. https://doi.org/10.1016/B978-0-444-63326-2.00003-X.
73. Hernandez C.C., Tian X.J., Hu N., et al. Dravet syndrome-associated mutations in GABRA1, GABRB2 and GABRG2 define the genetic landscape of defects of GABAA receptors. Brain Commun. 2021; 3 (2): fcab033. https://doi.org/10.1093/braincomms/fcab033.
74. Barker-Haliski M., White H.S. Glutamatergic mechanisms associated with seizures and epilepsy. Cold Spring Harb Perspect Med. 2015; 5 (8): a022863. https://doi.org/10.1101/cshperspect.a022863.
75. Zaitsev A.V., Smolensky I.V., Jorratt P., Ovsepian S.V. Neurobiology, functions, and relevance of excitatory amino acid transporters (EAATs) to treatment of refractory epilepsy. CNS Drugs. 2020; 34 (11): 1089–103. https://doi.org/10.1007/s40263-020-00764-y.
76. Lazarowski A., Czornyj L., Lubienieki F., et al. ABC transporters during epilepsy and mechanisms underlying multidrug resistance in refractory epilepsy. Epilepsia. 2007; 48 (Suppl. 5): 140–9. https://doi.org/10.1111/j.1528-1167.2007.01302.x.
77. Lazarowski A., Massaro M., Schteinschnaider A., et al. Neuronal MDR-1 gene expression and persistent low levels of anticonvulsants in a child with refractory epilepsy. Ther Drug Monit. 2004; 26 (1): 44–6. https://doi.org/10.1097/00007691-200402000-00010.
78. Dalaklioglu S. Evaluating appropriateness of digoxin, carbamazepine, valproic acid, and phenytoin usage by therapeutic drug monitoring. Clin Lab. 2013; 59 (3–4): 325–31. https://doi.org/10.7754/clin.lab.2012.120425.
79. Löscher W., Potschka H., Sisodiya S.M., Vezzani A. Drug resistance in epilepsy: clinical impact, potential mechanisms, and new innovative treatment options. Pharmacol Rev. 2020; 72 (3): 606–38. https://doi.org/10.1124/pr.120.019539.
80. Simon C., Stieger B., Kullak-Ublick G.A., et al. Intestinal expression of cytochrome P450 enzymes and ABC transporters and carbamazepine and phenytoin disposition. Acta Neurol Scand. 2007; 115 (4): 232–42. https://doi.org/10.1111/j.1600-0404.2006.00761.x.
81. van Vliet E.A., van Schaik R., Edelbroek P.M., et al. Region-specific overexpression of P-glycoprotein at the blood-brain barrier affects brain uptake of phenytoin in epileptic rats. J Pharmacol Exp Ther. 2007; 322 (1): 141–7. https://doi.org/10.1124/jpet.107.121178.
82. Löscher W., Luna-Tortós C., Römermann K., Fedrowitz M. Do ATP-binding cassette transporters cause pharmacoresistance in epilepsy? Problems and approaches in determining which antiepileptic drugs are affected. Curr Pharm Des. 2011; 17 (26): 2808–28. https://doi.org/10.2174/138161211797440212.
83. Fang M., Xi Z.Q., Wu Y., Wang X.F. A new hypothesis of drug refractory epilepsy: neural network hypothesis. Med Hypotheses. 2011; 76 (6): 871–6. https://doi.org/10.1016/j.mehy.2011.02.039.
84. Schmidt D., Löscher W. Drug resistance in epilepsy: putative neurobiologic and clinical mechanisms. Epilepsia. 2005; 46 (6): 858–77. https://doi.org/10.1111/J.1528-1167.2005.54904.x.
85. Bethmann K., Fritschy J.M., Brandt C., Löscher W. Antiepileptic drug resistant rats differ from drug responsive rats in GABA A receptor subunit expression in a model of temporal lobe epilepsy. Neurobiol Dis. 2008; 31 (2): 169–87. https://doi.org/10.1016/j.nbd.2008.01.005.
86. Volk H.A., Arabadzisz D., Fritschy J.M., et al. Antiepileptic drug-resistant rats differ from drug-responsive rats in hippocampal neurodegeneration and GABA(A) receptor ligand binding in a model of temporal lobe epilepsy. Neurobiol Dis. 2006; 21 (3): 633–46. https://doi.org/10.1016/j.nbd.2005.09.006.
87. Wiebe S., Jette N. Pharmacoresistance and the role of surgery in difficult to treat epilepsy. Nat Rev Neurol. 2012; 8 (12): 669–77. https://doi.org/10.1038/nrneurol.2012.181.
88. Hitiris N., Mohanraj R., Norrie J., et al. Predictors of pharmacoresistant epilepsy. Epilepsy Res. 2007; 75 (2–3): 192–6. https://doi.org/10.1016/j.eplepsyres.2007.06.003.
89. Mckee A.C., Daneshvar D.H. The neuropathology of traumatic brain injury. Handb Clin Neurol. 2015; 127: 45–66. https://doi.org/10.1016/B978-0-444-52892-6.00004-0.
90. Rogawski M.A. The intrinsic severity hypothesis of pharmacoresistance to antiepileptic drugs. Epilepsia. 2013; 54 (Suppl. 2): 33–40. https://doi.org/10.1111/epi.12182.
91. Sillanpää M., Schmidt D. Natural history of treated childhood-onset epilepsy: prospective, long-term population-based study. Brain. 2006; 129 (Pt 3): 617–24. https://doi.org/10.1093/brain/awh726.
92. Schmidt D.M., Löscher W.P. New developments in antiepileptic drug resistance: an integrative view. Epilepsy Curr. 2009; 9 (2): 47–52. https://doi.org/10.1111/J.1535-7511.2008.01289.x.
93. Löscher W., Schmidt D. Modern antiepileptic drug development has failed to deliver: ways out of the current dilemma. Epilepsia. 2011; 52 (4): 657–78. https://doi.org/10.1111/j.1528-1167.2011.03024.x.
94. Friedman A., Heinemann U. Role of blood-brain barrier dysfunction in epileptogenesis. In: Jasper's basic mechanisms of the epilepsies. 4th ed. Bethesda (MD): National Center for Biotechnology Information; 2012: 353–61. https://doi.org/10.1093/med/9780199746545.003.0027.
95. Panina Y.S., Timechko E.E., Usoltseva A.A., et al. Biomarkers of drug resistance in temporal lobe epilepsy in adults. Metabolites. 2023; 13 (1): 83. https://doi.org/10.3390/metabo13010083.
Якимов Алексей Михайлович.
ул. Партизана Железняка, д. 1, Красноярск, 660022
WoS ResearcherID AEW-9605-2022, Scopus Author ID 58161933100
Тимечко Елена Евгеньевна.
ул. Партизана Железняка, д. 1, Красноярск, 660022
WoS ResearcherID CAF-2677-2022
Парамонова Анастасия Ивановна.
ул. Партизана Железняка, д. 1, Красноярск, 660022
WoS ResearcherID HMP-3496-2023
Васильева Анастасия Александровна.
ул. Партизана Железняка, д. 1, Красноярск, 660022
Рыбаченко Фёдор Константинович.
ул. Партизана Железняка, д. 1, Красноярск, 660022
Рыбаченко Анастасия Дмитриевна.
ул. Партизана Железняка, д. 1, Красноярск, 660022
Дмитренко Диана Викторовна - д.м.н.
ул. Партизана Железняка, д. 1, Красноярск, 660022
WoS ResearcherID H-7787-2016
Якимов А.М., Тимечко Е.Е., Парамонова А.И., Васильева А.A., Рыбаченко Ф.К., Рыбаченко А.Д., Дмитренко Д.В. Гипотезы развития и стратегии преодоления фармакорезистентности при эпилепсии. Часть I: Гипотезы развития. Эпилепсия и пароксизмальные состояния. 2024;16(4):375-384. https://doi.org/10.17749/2077-8333/epi.par.con.2024.210
Yakimov A.M., Timechko E.E., Paramonova A.I., Vasilieva A.A., Rybachenko F.K., Rybachenko A.D., Dmitrenko D.V. Hypotheses of development and strategies for overcoming drug resistance in epilepsy. Part I: Hypotheses of development. Epilepsy and paroxysmal conditions. 2024;16(4):375-384. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2024.210

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































