


Содержание
Перейти к:
 Т. В. Кожанова,
С. С. Жилина,
Т. И. Мещерякова,
А. А. Абрамов,
М. М. Абидова,
Т. С. Каминская,
А. И. Крапивкин,
Н. Н. Заваденко
Т. В. Кожанова,
С. С. Жилина,
Т. И. Мещерякова,
А. А. Абрамов,
М. М. Абидова,
Т. С. Каминская,
А. И. Крапивкин,
Н. Н. Заваденко https://doi.org/10.17749/2077-8333/epi.par.con.2025.230
Перейти к:
Энцефалит в педиатрической практике – группа заболеваний, характеризующихся воспалением головного мозга, основными проявлениями которого являются лихорадка, судороги, плеоцитоз спинномозговой жидкости и нейрорадиологические изменения. Белки – деструкторы цитокинеза (англ. dedicator of cytokinesis protein, DOCK) имеют ведущее значение в регуляции актинового цитоскелета. DOCK11 играет важную роль в иммунных заболеваниях человека. В данной работе описан клинический случай анти-N-метил-D-аспартат-рецепторного энцефалита у мальчика с выявленным гемизиготным вариантом в гене DOCK11. Дефицит DOCK11 – это новая иммуноопосредованная актинопатия, которая связана с Х-хромосомой и вызывает нарушение активности белка 42, контролирующего клеточное деление (англ. cell division cycle 42, CDC42), и активацию преобразователя сигнала и активатора транскрипции 5 (англ. signal transducer and activator of transcription 5, STAT5). Она ассоциирована с аномальным ремоделированием актинового цитоскелета, а также фенотипом регуляторных Т-клеток, что приводит к нарушению регуляции иммунитета и тяжелому аутоиммунитету с ранним началом.
Кожанова Т.В., Жилина С.С., Мещерякова Т.И., Абрамов А.А., Абидова М.М., Каминская Т.С., Крапивкин А.И., Заваденко Н.Н. Новая Х-сцепленная иммуноопосредованная актинопатия у мальчика с анти-NMDA-рецепторным энцефалитом и вариантом в гене DOCK11. Эпилепсия и пароксизмальные состояния. 2025;17(2):170-181. https://doi.org/10.17749/2077-8333/epi.par.con.2025.230
Kozhanova T.V., Zhylina S.S., Meshcheryakova T.I., Abramov A.A., Abidova M.M., Kaminskaya T.S., Krapivkin A.I., Zavadenko N.N. A novel X-linked immune-mediated actinopathy in a boy with anti-NMDA receptor encephalitis and variant in DOCK11 gene. Epilepsy and paroxysmal conditions. 2025;17(2):170-181. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2025.230
Энцефалит в педиатрической практике – группа заболеваний, характеризующихся воспалением головного мозга, основными проявлениями которого являются лихорадка, судороги, плеоцитоз спинномозговой жидкости и нейрорадиологические изменения. Этиология энцефалита устанавливается только в половине случаев, причем наиболее распространенными являются вирусная и аутоиммунная [1]. В остальных случаях механизм его возникновения остается необъяснимым [1][2]. В научной литературе обсуждается генетическая предрасположенность к развитию энцефалита. В настоящее время благодаря совершенствованию технологии массового параллельного секвенирования идентифицирован ряд генов, варианты в которых приводят к дисфункции иммунной системы [3].
В основе нейровоспаления могут лежать изменения в геноме. Например, гемофагоцитарный лимфогистиоцитоз – это моногенное заболевание, характеризующееся нерегулируемым гипервоспалительным цитотоксическим ответом NK- и T-клеток, при котором вовлечение центральной нервной системы (ЦНС) является неблагоприятным прогностическим признаком [4]. При системной красной волчанке определенные локусы в генах TREX1, PTPN22, FCGR2A, FCGR2B, CTLA4 связаны с поражением ЦНС [5]. Кроме того, варианты в гене TLR3 ассоциированы с тяжелым герпетическим энцефалитом [6]. Таким образом, рядом автором предположено, что дети с необъяснимым энцефалитом могут быть носителями вариантов в генах, связанных с нейроиммунологическими состояниями, которые предрасполагают к воспалению ЦНС.
Иммуноопосредованные актинопатии представляют собой врожденные нарушения иммунитета, обусловленные дефектами генов, влияющими на ремоделирование актинового цитоскелета. Преобладающую роль в данной патологии играют представители семейства белков – деструкторов цитокинеза (англ. dedicator of cytokinesis protein, DOCK). В частности, описано участие белка DOCK11 в иммунных заболеваниях человека, но случаев выявления вариантов в гене DOCK11 у пациентов с аутоиммунным энцефалитом еще не установлено [7].
В данной работе мы описываем пациента с тяжелым развитием анти-N-метил-D-аспартат-рецепторного (англ. N-methyl-D-aspartate, NMDA) энцефалита и выявленным патогенным вариантов в гене DOCK11.
Пробанд, мальчик (возраст 1 год 8 мес), поступил в отделение реанимации и интенсивной терапии (ОРИТ) ГБУЗ «Научно-практический центр специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого Департамента здравоохранения г. Москвы» (ГБУЗ «НПЦ спец. мед. помощи детям ДЗМ») с диагнозом: «Некротизирующий энцефалит, спастический тетрапарез, судорожный синдром, двусторонний гнойный бронхит в анамнезе, острый инфекционный гастроэнтерит (среднетяжелая форма), токскикоз с эксикозом 1–2-й степени (реконвалесцент), острый назофарингит, гепатит неуточненной этиологии, синдром цитолиза и синдром системного воспалительного ответа инфекционного происхождения с органическим нарушением в анамнезе».
Пробанд от первой беременности в неродственном браке, протекавшей на фоне гестационного сахарного диабета (диетотерапия), острая респираторная вирусная инфекция на 12-й неделе (легкая форма). Роды на 40-й неделе, самостоятельные. Масса тела при рождении 4040 г, длина 53 см. Оценка по шкале Апгар 9/10 баллов. Выписан на 3-и сутки жизни. Грудное вскармливание до настоящего заболевания.
В раннем возрасте был спокойный. Хорошо ел, спал, прибавлял в массе. Моторное развитие: голову держит с 3 мес, переворачивается с 4 мес, сел в 8 мес, пополз в 8 мес, самостоятельно начал ходить с 1 года 2 мес. Речевое развитие: с 1 года появились отдельные слова, фразовой речи не было.
Дебют заболевания с 1 года 7 мес, когда появилась лихорадка до 38,5 °С, использован ибупрофен с хорошим эффектом. Утром был в сознании (играл, аппетит был сохранен), отмечался 2 раза жидкий стул, 1 раз рвота на высоте лихорадки, использован парацетамол без эффекта. Бригадой скорой медицинской помощи введена литическая смесь. Ребенок доставлен в ГБУЗ «Детская городская клиническая больница № 9 им. Г.Н. Сперанского ДЗМ» с диагнозом «острая кишечная инфекция». С момента поступления спал, в вечерние часы отмечено тоническое напряжение рук и ног, ребенок переведен в ОРИТ на 2-е сутки заболевания.
При поступлении в ОРИТ состояние тяжелое, уровень сознания – сопор, тоническое напряжение, гемодинамика стабильная, сатурации крови кислородом 99%. В динамике повышение уровня С-реактивного белка (СРБ) от 2,9 до 15 мг/л, прокальцитонин (ПКТ) 34,35 нг/мл. Ребенок переведен на искусственную вентиляцию легких, проведены седация, эмпирическая антибактериальная, противовирусная, инфузионная, гормональная, антикоагулянтная терапия. Учитывая данные анамнеза заболевания (острое развитие неврологической симптоматики), клинико-лабораторные данные (тонические судороги, нарушение цикла «бодрствование – сон», сонливость, в ликворограмме цитоза нет (3 кл), белок повышен до 1 г/л (белково-клеточная диссоциация), синдром цитолиза – аланинаминотрансфераза (АЛТ) 1219,6 Ед/л, аспартатаминотрансфераза (АСТ) 2600 Ед/л, умеренная гипераммониемия 102,39 мкмоль/л), данные инструментальных обследований (на компьютерной томографии головного мозга – очаги ишемии в перивентрикулярных областях, на магнитно-резонансной томографии (МРТ) головного мозга с контрастированием выявлены участки патологического МР-сигнала в паренхиме больших полушарий и мозжечка), установлен диагноз «некротизирующий энцефалит».
В течение 1-х суток в ОРИТ была начата терапия внутривенными иммуноглобулинами (1 г/кг) – габриглобин 12,5 г, гормональная терапия дексаметазоном 0,6 мг/кг/сут в 3 введения внутривенно (в/в) струйно, антибактериальная терапия – меропенем 120 мг/кг/сут в/в капельно в 3 введения, линезолид 10 мг/кг в/в капельно каждые 8 ч, противогерпетическая терапия – ацикловир 500 мг/м² каждые 8 ч в/в капельно), цитофлавин в/в, продленная седация мидазоламом. Тесты полимеразной цепной реакции (ПЦР) на респираторные вирусы и вирусы гриппа отрицательные. Посев ликвора стерилен. ПЦР-диагностика крови на вирус простого герпеса 6 (лат. herpes simplex virus 6 , HSV-6): обнаружено ++lg572.
В дальнейшем после отмены медицинской седации у ребенка отмечались эпизоды тонических судорог, в связи с чем проводилась противоэпилептическая терапия вальпроевой кислотой (сначала в/в капельно, далее пероральное введение 30 мг/кг/сут), паглюфералом-2 (0,5 таблетки 2 раза в сутки). На фоне проводимой терапии отмечена положительная динамика, выполнены экстубация трахеи и перевод на самостоятельное дыхание, оксигенотерапия проводилась менее 1 сут.
На 18-е сутки в ОРИТ дыхание самостоятельное без дотации кислорода, респираторно ребенок компенсирован, неврологически сохраняется церебральная недостаточность до 11 баллов по шкале комы Глазго (ШКГ), на тактильные раздражители реагирует открыванием глаз, отдергиванием конечностей, взор не фиксирует, двигательной активности нет, спастический тетрапарез. Гемодинамика стабильна. Энтерально кормится. Диурез сохранен. Стул без патологических примесей. Уровень аминотрансфераз снизился: АЛТ 201 Ед/л, АСТ 101 Ед/л, уровень аммония без существенной динамики (108,0 мкмоль/л). Маркеры инфекционно-воспалительного процесса отрицательные (СРБ 0 мг/л, ПКТ менее 0,05 нг/мл). На фоне проводимого лечения отмечено снижение вирусной нагрузки – ДНК HSV-6 слабоположительна (+lg5,4). При серологическом исследовании выявлены антитела иммуногробулина G (англ. immunoglobulin G, IgG) к цитомегаловирусу, вирусу Эпштейна–Барр, HSV-1/2 при отсутствии IgM, что в совокупности с отсутствием ДНК указанных вирусов в ликворе и крови позволяет исключить активную инфекцию. На 24-е сутки ребенок переведен на долечивание в НПЦ спец. мед. помощи детям ДЗМ.
При поступлении в ОРИТ НПЦ спец. мед. помощи детям ДЗМ состояние пациента было тяжелым вследствие неврологической симптоматики, обусловленной основным заболеванием. Ребенок на самостоятельном дыхании, без дотации кислорода.
Неврологический статус
Сознание: глубокое оглушение. Положение вынужденное на спине. Конституция нормостеническая. Питание удовлетворительное. ШКГ 11 баллов. По педиатрической шкале последовательной оценки органной недостаточности (англ. рediatric Sequential Organ Failure Assessment, pSOFA) судорог не отмечалось. Лицевых и скелетных дизморфий нет. Глаза полуприкрыты. Взгляд кратковременно фиксирует. Не следит. Ресничный, корнеальные рефлексы вызываются. Мимических реакций нет. Кормится через зонд. Периодически стонет и делает сосательные движения. Спонтанной и целенаправленной двигательной активности нет. Руки в положении разгибания, кисти «когтистые». Дистония. Тугоподвижность в суставах. Кожа сухая.
В неврологическом статусе уровень сознания – оглушение, гипокинетический мутизм, комплексная моторная афазия, идеомоторная апраксия, апраксия глотания. Оромандибулярная дискинезия. Асимметричный тетрапарез. Псевдобульбарный синдром.
Данные МРТ от 05.03.2024 г.
МРТ головного и спинного мозга с внутривенным контрастированием выполнена 05.03.2024 г. на аппарате MEXL-3010/G5 (Toshiba, Япония) с индукцией магнитного поля 3 Тл со сверхпроводящим магнитом.
При сравнении с результатами ранее проведенной МРТ на фоне уменьшения объема мозгового вещества ранее выявленных зон патологического изменения МР-сигнала от перивентрикулярного и глубинного белого вещества лобно-теменных областей больших полушарий, таламусов, ножек среднего мозга и расширения периваскулярных пространств в семиовальных центрах и базальных ядрах, белом веществе гемисфер и червя мозжечка участки с признаками ограничения диффузии в указанных структурах головного мозга уменьшились по размерам и степени рестрикции диффузии. На фоне появления участков кровоизлияний в таламусах и линейных диапедезных кровоизлияний в глубинном белом веществе гемисфер мозжечка участков патологического накопления контраста в структурах головного мозга не выявлено. Размеры всех отделов желудочковой системы увеличились. Умеренно увеличились конвекситальные скопления ликвора в расширенных щелях и бороздах больших полушарий. Парастволовые и церебелломедулярная цистерны расширены в большей степени.
Заключение: «МР-картина формирования глиозно-атрофических изменений в структурах головного мозга с геморрагическим компонентом в таламусах и гемисферах мозжечка как этап течения некротизирующего энцефалита (с учетом анамнеза). Отрицательная динамика в виде заместительного расширения желудочковой системы, субарахноидальных и цистернальных пространств. Патологии позвоночника и спинного мозга не выявлено» (рис. 1).
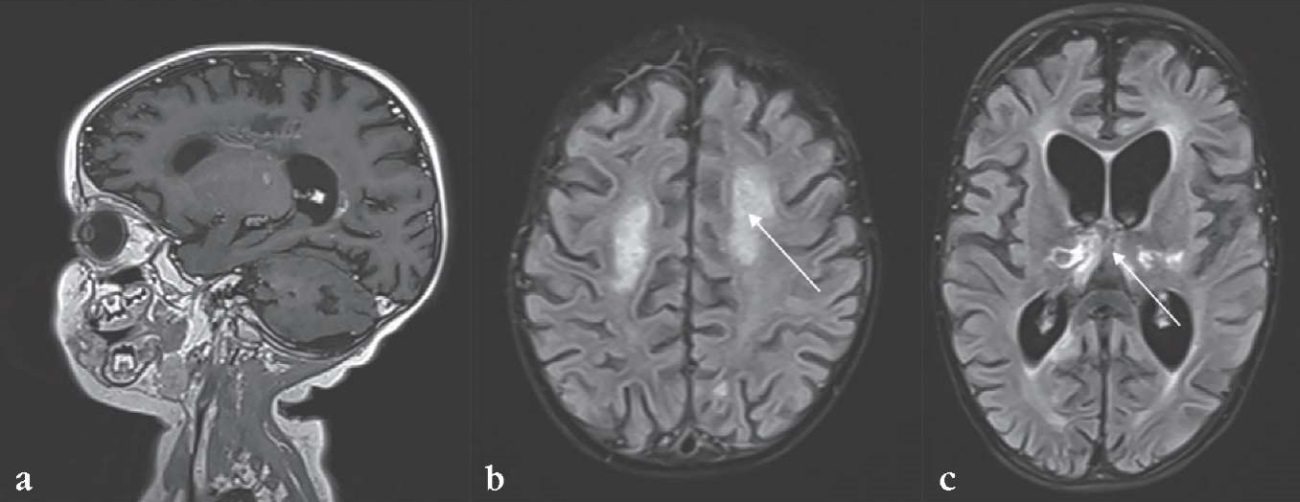
Рисунок 1. Результаты магнитно-резонансной томографии пробанда от 05.03.2024 г. Картина формирования глиозно-атрофических изменений в структурах головного мозга с геморрагическим компонентом в таламусах и гемисферах мозжечка:
а – уменьшение объема мозгового вещества, вентрикуломегалия без нарушения оттока ликвора; b – глиозно-атрофические изменения в структурах головного мозга (стрелка); с – участки кровоизлияний в таламусах и линейные диапедезные кровоизлияния в глубинном белом веществе (стрелка)
Figure 1. Proband magnetic resonance imaging (05.03.2024). Formation of glial-atrophic changes in brain structures with hemorrhagic alteration in the thalami and cerebellar hemispheres:
a – decreased volume of brain substance, ventriculomegaly without altered cerebrospinal fluid outflow; b – glial-atrophic changes in brain structures (arrow); c – areas of hemorrhage in the thalami and linear diapedetic hemorrhages in the deep white matter (arrow)
Люмбальная пункция
При проведении люмбальной пункции цитоз, белок, глюкоза в пределах референсных значений.
Данные видео-ЭЭГ-мониторинга с записью сна от 05.03.2024 г.
Электроэнцефалографический (ЭЭГ) видеомониторинг выполнен 05.03.2024 г. на модульной нейродиагностической системе NicoletOne (VIASYS Healthcare, США).
Корковая ритмика представлена диффузной дельта-активностью амплитудой 100–150 мкВ без зональных различий и регистрации основных ритмов в сочетании с мышечными артефактами (вероятно, в состоянии бодрствования). Эпилептиформной активности, эпилептических приступов и их ЭЭГ-паттернов не зарегистрировано (рис. 2).
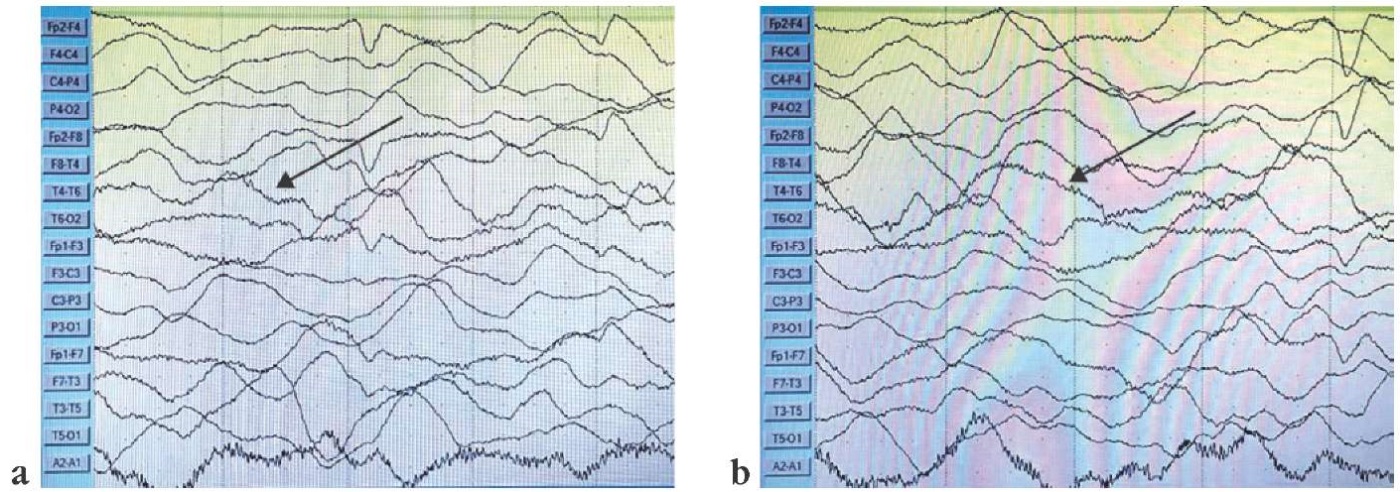
Рисунок 2. Результаты электроэнцефалографического видеомониторинга пробанда от 05.03.2024 г. Корковая ритмика представлена диффузной дельта-активностью амплитудой 100–150 мкВ без зональных различий и регистрации основных ритмов в сочетании с мышечными артефактами. Стрелками указана «экстремальная дельта-щетка»:
а – диффузная дельта-активность; b – «экстремальная дельта-щетка»
Figure 2. Proband electroencephalographic video monitoring (05.03.2024). Cortical rhythms are represented as diffuse delta activity with 100–150 μV amplitude without zonal differences and recorded main rhythms in combination with muscle artifacts. Arrows indicate the “extreme delta brush”:
a – diffuse delta activity; b – “extreme delta brush”
На фоне проводимого лечения уровень сознания максимально 13 баллов по ШКГ. Для дальнейшего лечения пробанд переведен в психоневрологическое отделение № 2 НПЦ спец. мед. помощи детям ДЗМ.
Неврологический статус
В неврологическом статусе кратковременная фиксация взора, косоглазия нет, фотореакции сохранены. Нистагма нет, нёбные и глоточные рефлексы вызываются, uvula по средней линии, язык по средней линии в полости рта. Объем пассивных движений полный. Эквинусная установка голеностопных суставов. Сгибательная установка локтевых суставов. Мышечный тонус повышен по спастическому типу, D=S. Сухожильные рефлексы средней степени в руках, оживлены в ногах, D=S. Положительный симптом Бабинского с двух сторон. Кожные рефлексы снижены. Целенаправленных движений нет. На боль реагирует плачем. Психомоторное развитие: голову не удерживает, не переворачивается, не садится самостоятельно, не сидит, не встает, не ходит. Речи нет.
Лабораторные исследования
28.03.2024 г. получен положительный результат анализа крови на аутоиммунные антитела (anti-NMDA 1:100). Проведено исследование антител к вирусным инфекциям (метод парных сывороток – сравнение нарастания титра в динамике): нарастания титра антител класса G к цитомегаловирусу (26.02.2024 г. – 132,3 МЕ/мл, 22.03.2024 г. – 45,1 МЕ/мл), к вирусу простого герпеса (26.02.2024 г. – 29,6 МЕ/мл, 22.03.2024 г. – 14,23 МЕ/мл) не выявлено. Клинический анализ ликвора от 22.03.2024 г. в пределах референсных значений. Данные клинического анализа крови от 12.03.2024 г. без значимых отклонений.
МРТ головного мозга от 03.04.2024 г.
При сравнении результатов МРТ головного мозга с внутривенным контрастированием с данными от 05.03.2024 г. определяется уменьшение объема мозгового вещества в зонах патологического изменения МР-сигнала от перивентрикулярного и глубинного белого вещества лобно-теменных областей больших полушарий, таламусов, ножек среднего мозга, в белом веществе гемисфер и червя мозжечка, отмечается появление разнокалиберных дефектов в перивентрикулярном и глубинном белом веществе лобно-теменных областей, таламусах и гемисферах мозжечка. На фоне инволюционирующего геморрагического компонента в таламусах определяется фрагментарное линейное накопление контраста по контурам формирующихся дефектов мозгового вещества в белом веществе больших полушарий, таламусах и глубинном белом веществе гемисфер мозжечка (рис. 3).
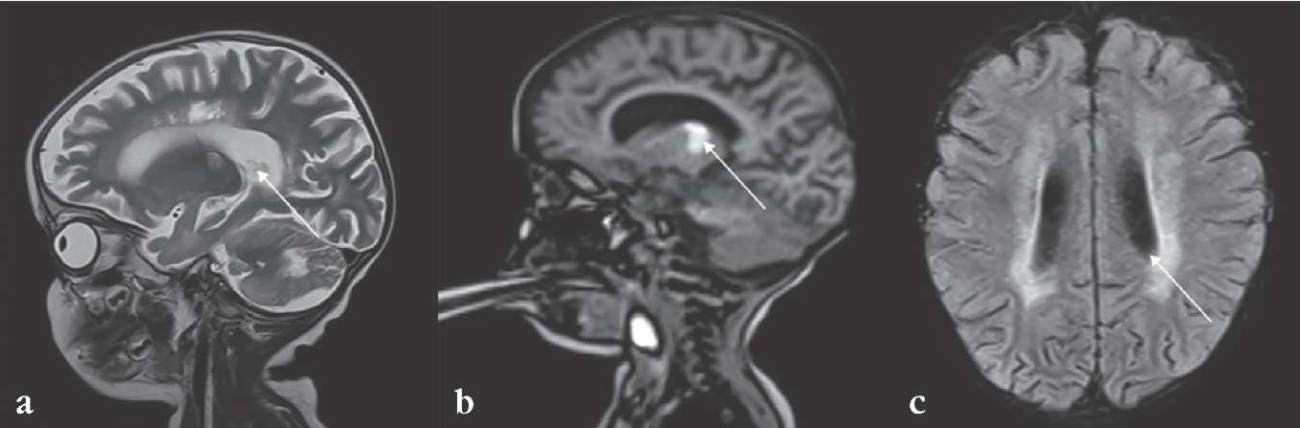
Рисунок 3. Результаты магнитно-резонансной томографии пробанда от 03.04.2024 г. В динамике нарастание атрофии и появление участков кистозной трансформации в больших полушариях, таламусах и гемисферах мозжечка. Увеличение заместительного расширения желудочковой системы, субарахноидальных и цистернальных пространств:
а – уменьшение объема мозгового вещества в зонах патологического изменения магнитно-резонансного сигнала от перивентрикулярного и глубинного белого вещества лобно-теменных областей больших полушарий, таламусов, ножек среднего мозга, в белом веществе гемисфер и червя мозжечка, отмечается появление разнокалиберных дефектов в перивентрикулярном и глубинном белом веществе лобно-теменных областей, таламусах и гемисферах мозжечка (стрелка); b – в таламусах определяется фрагментарное линейное накопление контраста по контурам формирующихся дефектов мозгового вещества в белом веществе больших полушарий, в таламусах и глубинном белом веществе гемисфер мозжечка (стрелка); c – расширение желудочковой системы (стрелка)
Figure 3. Proband magnetic resonance imaging (03.04.2024). Dynamically extending atrophy and emerging areas of cystic transformation in cerebral hemispheres, thalami and cerebellar hemispheres. Enlarged substitution expansion of the ventricular system, subarachnoid and cisternal spaces:
a – decreased volume of brain matter in the areas of pathological change in the magnetic resonance signal from periventricular and deep white matter of frontal-parietal regions of cerebral hemispheres, thalami, midbrain peduncles, in the white matter of hemispheres and cerebellar vermis, emergence of different-sized defects is noted in periventricular and deep white matter of frontal-parietal regions, thalami and cerebellar hemispheres (arrow); b – in the thalami, fragmentary linear contrast accumulation is observed along the contours of developing defects in the white matter of cerebral hemispheres, thalami and deep white matter of cerebellar hemispheres (arrow); c – ventricular system expansion (arrow)
Заключение: «МР-картина глиозно-атрофических изменений структур головного мозга с геморрагическим компонентом как следствие течения некротизирующего энцефалита (с учетом анамнеза), нарастание атрофии и появление участков кистозной трансформации в больших полушариях, таламусах и гемисферах мозжечка. Увеличение заместительного расширения желудочковой системы, субарахноидальных и цистернальных пространств».
Установлен диагноз: «Другой энцефалит, миелит и энцефаломиелит. Анти-NMDA-энцефалит. Гипоксически-геморрагическое поражение головного мозга с формированием глиозно-атрофических изменений в структурах головного мозга с геморрагическим компонентом в таламусах и гемисферах мозжечка. Энцефалопатия тяжелой степени, гипокинетический мутизм, моторная афазия, апраксия глотания, псевдобульбарный синдром (G04.8). Клиническое осложнение основного диагноза: судорожный синдром в стадии медикаментозной ремиссии (R56.8). Клинический сопутствующий диагноз: токсическое поражение печени с картиной гепатита, не классифицированное в других рубриках (K71.6)».
Учитывая отрицательную динамику по данным МРТ, накопление контраста как признак продолжающегося воспалительного процесса, несмотря на проведенные курсы терапии глюкокортикостероидом 0,6 мг/кг/сут, двукратное введение иммуноглобулинов в дозе 1 г/кг/сут, эффект от проводимой терапии следует расценивать как недостаточный. Ребенку показано назначение терапии препаратами второй линии – ритуксимаб из расчета 750 мг/м² (400 мг в/в, повторное введение через 2 нед).
Данные видео-ЭЭГ-мониторинга с записью сна от 20.05.2024 г.
Выраженные диффузные изменения корковой ритмики. При проведении функциональных проб значимых изменений корковой ритмики не зарегистрировано. Дифференцировка сна на стадии сглажена. В бодрствовании и во сне регистрируется в структуре продолженного замедления региональная эпилептиформная активность: в правой центральной области – в виде одиночных и сгруппированных комплексов «пик – медленная волна» амплитудой до 180 мкВ с тенденцией к диффузному распространению, в левой лобно-центральной области – в виде одиночных и сгруппированных комплексов «пик – медленная волна» амплитудой до 400 мкВ с тенденцией к диффузному распространению, в левой центральной области – в виде одиночных и сгруппированных комплексов «пик – медленная волна» амплитудой до 130 мкВ, в передних вертексных отделах – в виде одиночных и сгруппированных комплексов «пик – медленная волна» амплитудой до 120 мкВ. Индекс эпилептиформной активности варьируется от средних до высоких значений (50–60%). Эпилептических приступов и их ЭЭГ-паттернов за время исследования не зарегистрировано (рис. 4).
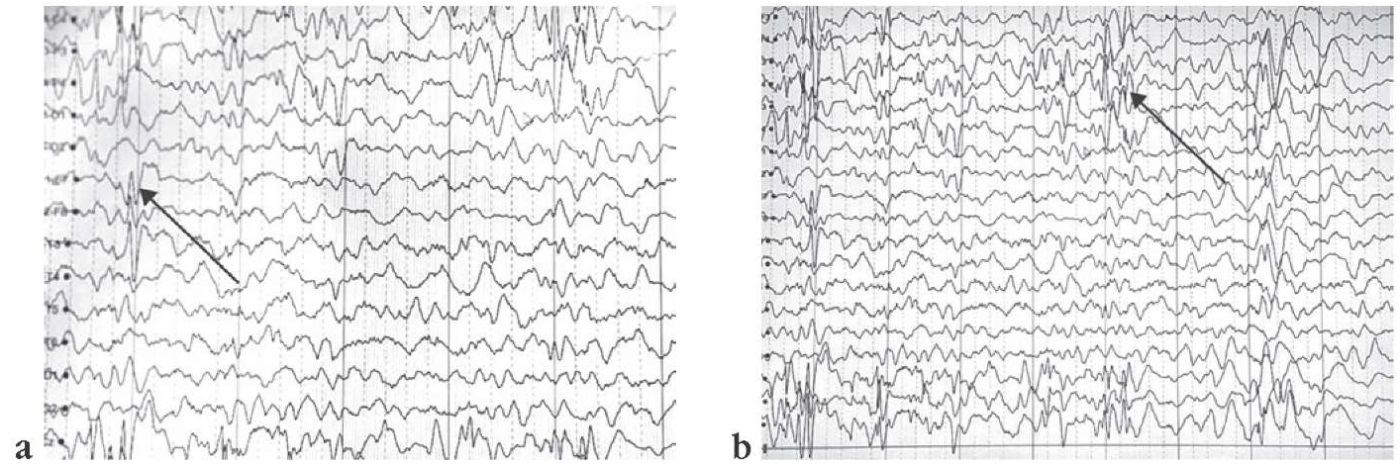
Рисунок 4. Результаты электроэнцефалографического видеомониторинга пробанда от 20.05 2024 г.:
а – во сне регистрируется в структуре продолженного замедления региональная эпилептиформная активность в правой центральной области (стрелка); b – в передних вертексных отделах одиночные и сгруппированные комплексы «пик – медленная волна» амплитудой до 120 мкВ (стрелка)
Figure 4. Proband electroencephalographic video monitoring (20.05.2024):
a – regional epileptiform activity in sleep is recorded in the pattern of prolonged slowing in the right central region (arrow); b – in anterior vertex regions, single and grouped peak-slow wave complexes with amplitude of up to 120 μV (arrow)
Терапия
На фоне лечения отмечено уменьшение спастичности в нижних и правой верхней конечностях, фотореакции улучшились. Ребенок стал фиксировать и прослеживать взглядом за предметами, появились минимальные движения в конечностях, движения головой в стороны в горизонтальном направлении, стал недлительно удерживать голову в вертикальном положении, купирована лихорадка. В течение госпитализации проведена коррекция медикаментозной терапии – с постепенным снижением до полной отмены баклофена, паглюферала, гидроксизина.
Ребенок повторно госпитализировался в августе 2024 г. Положительной динамики в неврологическом состоянии нет.
Консультация иммунолога: вторичное иммунодефицитное состояние. В связи с высоким риском инфекционных осложнений на фоне нарушения специфического синтеза антител по жизненным показаниям ребенок нуждается в заместительной терапии препаратом иммуноглобулина человека нормального в дозе 0,5 г/кг/сут 1 раз в 4 нед.
Согласно международным рекомендациям М. Nosadini et al. [8] ребенку с перенесенным анти-NMDA-энцефалитом от 02.2024 г. показано введение внутривенных иммуноглобулинов из расчета 1 г/кг в/в 1 раз в 4 нед (учитывая тяжесть и течение заболевания, возраст, рекомендации неврологов). Выбор препарата иммуноглобулина человека – в пользу препаратов, прошедших вирусинактивацию в цикле своего производства и с наличием скрининга на отсутствие в них вируса В19 для снижения риска тяжелых апластических состояний у пациентов с компрометированной иммунной системой [8]. После проведения врачебного консилиума на основании принятого им решения 17.08.2024 г. выполнена инфузия иммуноглобулина (иммуноглобулин человека нормальный) в объеме 200 мл (суммарная доза 10 г) без нежелательных реакций [8].
Данные МРТ от августа 2024 г.
Картина глиозно-атрофических изменений структур головного мозга с участками кистозной трансформации в лобно-теменных областях, таламусах и гемисферах мозжечка как следствие перенесенного некротизирующего энцефалита (с учетом анамнеза), без динамики. Небольшое уменьшение заместительного расширения желудочковой системы, субарахноидальных пространств.
Повторная госпитализация в ноябре 2024 г.
Положительная динамика в неврологическом статусе в виде улучшения зрительного, эмоционального и тактильного контакта. Ребенок кормится через назогастральный зонд. Речи нет. Объем пассивных движений полный. Целенаправленные движения бедные. Сгибательная установка локтевых суставов с пронацией предплечий. Приведение большого пальца правой кисти. Мышечный тонус дистоничный, D<S. Эквинусная установка голеностопных суставов. Сухожильные рефлексы средней степени в руках, оживлены в ногах, D=S. Положительный симптом Бабинского с двух сторон. Клонусы обеих стоп. Кожные рефлексы вызываются. За игрушкой тянется с дисметрией в рамках пареза. Больше манипулирует левой рукой. Психомоторное развитие: голову удерживает длительно, не переворачивается, не садится самостоятельно, не сидит посаженный, не встает, не ходит.
Логопедический статус: лицо симметрично. Мягкое нёбо подвижное. Uvula по средней линии. Язык по средней линии. Объем движений языка ограничен, тонус повышен. Глоточные рефлексы живые. Глотание нарушено, питание через зонд, парез надгортанника. Жевание нарушено. Дыхание свободное. Фонационный выдох укороченный. Вербальный контакт с использованием невербальных средств общения. Спонтанная речь: звукокомплексы. Автоматизированная речь невозможна. Повторная речь невозможна. Называние невозможно. Понимание ситуативных вопросов: понимает. Выполнение простых инструкций: выполняет. Выполнение развернутых инструкций: попытки выполнения. Показ предметных изображений: показывает верно. Оральный праксис: норма. Артикуляторный праксис: диспраксия. Сумма: 23. Результат: выявлены нарушения речи и когнитивных функций разной степени выраженности. Дисфагия легкой степени. Задержка речевого и познавательного развития вследствие перенесенного воспалительного заболевания ЦНС. Дисфагия.
Учитывая отсутствие подтверждающих вирусный энцефалит серологических маркеров, проводилось полноэкзомное секвенирование с целью выявления генетической природы заболеваний с энцефалитоподобным дебютом.
Полноэкзомное секвенирование выполнено в генетической лаборатории ГБУЗ «НПЦ спец. мед. помощи детям ДЗМ». Геномная ДНК выделена методом лизиса клеток с последующей очисткой на стекловолоконных фильтрах (реактивы QIAamp DNA Mini Kit – Qiagen, Германия), затем использована для приготовления геномных библиотек для массового параллельного секвенирования (NEBNext Ultra II – New England BioLabs, США). Из полученных библиотек методом гибридизации были отобраны только те участки ДНК, которые соответствуют экзонам генов и сайтам сплайсинга (SureSelect AllExon V7 – Agilent, США). Далее проведено определение их нуклеотидной последовательности на секвенаторе HiSeq 1500 (Illumina, США), реактивы HiSeq Rapid SBS Kit v2 (Illumina, США).
Выявлен ранее не описанный вариант нуклеотидной последовательности в экзоне 41/53 гена DOCK11 в гемизиготном состоянии (chrX:118649040; c.4494G>C, p.Arg1498Ser; NM_144658.4). Варианты нуклеотидной последовательности в гене DOCK11 в гемизиготном состоянии описаны у пациентов с «мультисистемным аутовоспалительным заболеванием, с иммунной дисрегуляцией, Х-сцепленным» (англ. аutoinflammatory disease, multisystem, with immune dysregulation, X-linked; MIM 301109), что предполагает X-сцепленный рецессивный тип наследования. Частота такого варианта нуклеотидной последовательности в контрольной выборке gnomAD в гетерозиготном состоянии составляет 0,008%, в европейской популяции – 0%. Выявленный вариант нуклеотидной последовательности не зарегистрирован в российской выборке RUSeq. Алгоритмы предсказания патогенности расценивают его как «вероятно патогенный». Данный вариант валидирован у пробанда методом секвенирования по Сэнгеру, исследовано его происхождение (сегрегационный анализ). У матери пробанда вариант в гене DOCK11 определен в гетерозиготном состоянии (рис. 5).
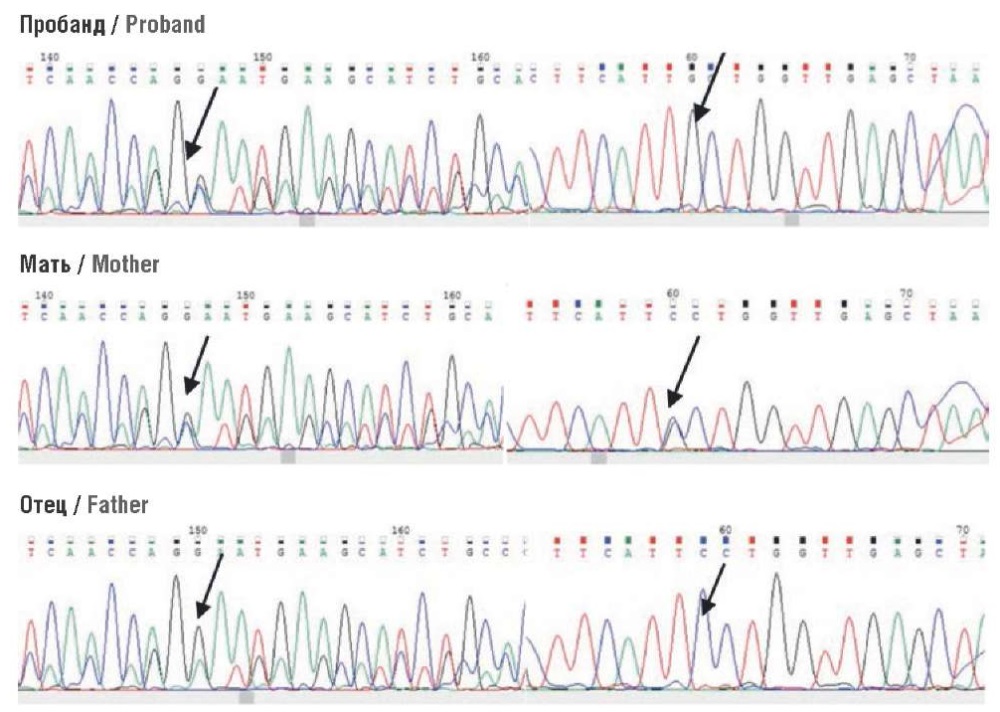
Рисунок 5. Результаты секвенирования по Сэнгеру 41-го экзона гена DOCK11 с прямым и обратным праймерами. Стрелками указана подтвержденная мутация у пробанда (патогенный аллель), выявленная мутация у матери (патогенный аллель) и ее отсутствие у отца (нормальный аллель)
Figure 5. Sanger sequencing of exon 41 of the DOCK11 gene with forward and reverse primers. Arrows indicate verified mutation in the proband (pathogenic allele), the detected mutation in the mother (pathogenic allele) and its absence in the father (normal allele)
Аутоиммунные энцефалиты в педиатрической практике имеют особую значимость в связи с высокими показателями заболеваемости и смертности, при этом этиология около 50% случаев остается неизвестной. Определенную роль в развитии энцефалитов играет генетическая предрасположенность, однако варианты в генах, которые могут приводить к данной патологии, полноценно не изучены.
D. Malik et al. [9] описали ребенка с анти-NMDA-энцефалитом и 10 детей с энцефалитом без установленной этиологии. Всем пациентам было выполнено полногеномное секвенирование с целью поиска вариантов нуклеотидной последовательности в генах, ассоциированных с моногенными наследственными нарушениями иммунной и нервной систем, а также полиморфизмов с повышенным риском развития данной группы заболеваний. Используя базу данных HGMD, авторы разделили выявленные варианты на следующие группы: моногенные патогенные и вероятно патогенные варианты и полиморфизмы, связанные с заболеваниями. У 6 (55%) пациентов обнаружены патогенные и вероятно патогенные варианты в генах, 5 (45%) были гетерозиготными носителями аутосомно-рецессивных заболеваний и 6 (55%) – носителями полиморфизмов. Среди обследованных детей 7 (64%) имели более одного варианта в генах, предполагающих возможный полигенный риск. В их число входили варианты, ассоциированные с развитием атипичного уремического синдрома, общих иммунодефицитных состояний, гемофагоцитарного лимфогистиоцитоза и системной красной волчанки. Таким образом, в данном исследовании показана роль вариантов в генах, ассоциированных с врожденными нарушениями иммунитета, в развитии энцефалитов у детей [9].
Врожденные дефекты иммунитета, или первичные иммунодефициты, – разнообразная группа генетической патологии, которая приводит к повышенной восприимчивости к инфекциям и может реализоваться такими иммунопатологическими процессами, как аутоиммунные, аутовоспалительные и (или) онкологические, реже – аллергические. В 2022 г. Международный союз иммунологических обществ (англ. International Union of Immunological Societies, IUIS) представил обновленную классификацию врожденных ошибок иммунитета [10], которая включает 485 заболеваний. В ней выделяется десять групп врожденных ошибок иммунитета:
1) комбинированные иммунодефициты (иммунодефициты с поражением клеточного и гуморального иммунитета);
2) комбинированные иммунодефициты, ассоциированные с синдромальными проявлениями (иммунодефициты с врожденной тромбоцитопенией – например, синдром Вискотта–Олдрича), многочисленные дефекты репарации ДНК (атаксия-телеангиоэктазия, синдром Ниймеген и пр.), иммунокостные дисплазии (синдром Шимке и пр.), иммунодефициты с дефектом тимуса (синдром Ди Джорджи, CHARGE), подгруппа синдромов гипериммуноглобулинемии Е);
3) преимущественно антительные дефекты, подразделяющиеся на агаммаглобулинемии, синдромы с фенотипом общей вариабельной иммунной недостаточности и различные другие селективные дефициты антител;
4) болезни иммунодисрегуляции (включают подгруппу гемофагоцитарных лимфогистиоцитозов);
5) врожденные дефекты числа и функций фагоцитов;
6) дефекты внутреннего и врожденного иммунитета (разнообразные моногенные синдромы, повышающие восприимчивость к разнообразным инфекциям);
7) различные аутовоспалительные синдромы, в патогенезе которых ключевым звеном является нарушение молекулярных механизмов регуляции воспаления;
8) дефекты комплемента;
9) различные типы анемии Фанкони, врожденные дискератозы и синдромы костно-мозговой недостаточности;
10) фенокопии первичных иммунодефицитов.
Для всех форм врожденных дефектов иммунитета установлена своя молекулярная основа.
Анти-NMDA-энцефалит – это аутоиммунный энцефалит, обусловленный увеличением уровня аутоантител IgG к субъединице NR1 рецептора NMDA. Клиническая картина анти-NMDA-энцефалита впервые описана R. Vitaliani et al. в 2005 г. [11]. В ней наблюдаются гриппоподобные проявления (повышение температуры, общая слабость, недомогание, боль в мышцах и суставах, головные боли и др.), вегетативные симптомы (тахикардия, повышение артериального давления и температуры, саливация, центральная гиповентиляция), неврологические расстройства (с постепенным развитием когнитивных нарушений, памяти и речи, двигательных расстройств, мозжечковой атаксии, снижения уровня сознания), аффективная симптоматика (как депрессивного, так и маниакального полюсов), психопатологические расстройства, которые зачастую неотличимы от симптоматики, наблюдаемой при шизофрении (бредовые идеи, зрительные и слуховые галлюцинации, кататония), нарушения поведения (психомоторное возбуждение, агрессия) и судороги.
Достоверно установлено, что иммунологическими триггерами анти-NMDA-энцефалита являются новообразования и герпетический энцефалит [12]. У 50% пациентов иммунологические триггеры анти-NMDA-энцефалита не выявляются [13]. Отсутствуют сведения по генетической предрасположенности к анти-NMDA-энцефалиту у детей. По данным, собранным к 2019 г., оценочная заболеваемость анти-NMDA-энцефалитом составляет 1,5 случая на 1 млн человек в год, при этом доля пациентов женского пола достигает примерно 80%, доля пациентов моложе 18 лет – примерно 37% [8][14]. Отсюда и попытки поиска генетических маркеров предрасположенности к аутоиммунным заболеваниям.
Множество неврологических, сосудистых и иммунных заболеваний человека, а также рак связаны, в частности, с аберрантным гомеостазом актинового цитоскелета [15][16]. К ним относятся иммуноопосредованные актинопатии, обусловленные дефектами генов, влияющих на ремоделирование актинового цитоскелета. На данный момент описано более 20 видов нарушений [15][17].
Ремоделирование актинового цитоскелета – это динамичный и строго регулируемый процесс, который имеет решающее значение для многочисленных клеточных процессов, включая морфологические изменения, миграцию, межклеточное взаимодействие и передачу сигналов, необходимые для развития и функционирования иммунных клеток [17]. Rho-гуанозинтрифосфатазы (Rho-ГТФазы) – семейство клеточных сигнальных белков, «малых» (около 21 кДа) G-белков. относящихся к суперсемейству Ras. К ним относятся три наиболее изученных белка RAC1, CDC42 и RHO, которые играют ключевую роль во внутриклеточной реорганизации актина [18]. Фенотипический спектр мутаций в гене RAC варьируется от дефицита гранулоцитов (англ. loss-of-function) до тяжелого комбинированного иммунодефицита с гипоплазией костного мозга (англ. gain-of-function) [19]. Варианты нуклеотидной последовательности de novo в гене CDC42 были недавно идентифицированы у пациентов с синдромом Такенучи–Косаки с сопутствующими признаками, такими как макротромбоцитопения и аномалии развития, или с неонатальной цитопенией с дисгематопоэзом, аутовоспалением, сыпью и гемофагоцитарным лимфогистиоцитозом (синдром NOCARH) [20]. Rho-ГТФазы активируются факторами обмена гуанина (англ. guanine exchange factors, GEFs), такими как члены семейства дедикаторов цитокинеза DOCK (DOCK2 и DOCK8). Значимость DOCK для иммунитета человека была проиллюстрирована на примере мутаций зародышевой линии в генах DOCK2 и DOCK8, лежащих в основе комбинированных иммунодефицитов с тяжелыми и рецидивирующими инфекциями, а также аутоиммунных проявлений, включая тромбоцитопению, гемолитическую анемию и васкулит. Биаллельные мутации в гене DOCK2 приводят к ранним тяжелым инвазивным бактериальным и вирусным инфекциям и Т-клеточной лимфопении. Варианты в гене DOCK8 – причина тяжелых бактериальных, вирусных и грибковых инфекций, а также экземы и выраженных аллергических реакций на окружающую среду. Преобладающую роль в иммуноопосредованных актинопатиях играют представители семейства белков DOCK. В частности, описано участие белка DOCK11 в иммунных заболеваниях человека [7].
Как и в случае с DOCK2 и DOCK8, DOCK11 преимущественно экспрессируется в кроветворных клетках, а у мышей он играет роль в раннем развитии и функционировании В-клеток. Однако его функция в биологии и заболеваниях иммунных клеток человека остается неизвестной. Дефицит DOCK11 может приводить к нарушению иммунной дисрегуляции у пациентов с восприимчивостью к инфекции, системному воспалению и нормоцитарной анемии [21].
J. Block et al. (2023 г.) [22] выявили редкие гемизиготные варианты loss-of-function в гене DOCK11, приводящие к нарушению активации CDC42, как причину новой формы иммунной дисрегуляции. Клинически дефицит DOCK11 включает фенотипы, связанные с вариантами в гене CDC42, такие как рецидивирующие инфекции, иммунная дисрегуляция, анемия, аномалии тромбоцитов и развития нервной системы. В отличие от пациентов, несущих варианты в гене CDC42, пациенты с дефицитом DOCK11 не имели дисморфологических черт лица, нейтропении, моноцитопении или гемофагоцитарного лимфогистиоцитоза. Несмотря на системный воспалительный фенотип при дефиците DOCK11, авторы не обнаружили аутоантител, изменений в количестве регуляторных Т-клеток или явных дефектов в сборке иммунных синапсов – признаков, которые наблюдаются при других генетических нарушениях, связанных с аберрантной сборкой актина. Примечательно, что у пациентов с дефицитом DOCK11 наблюдался высокий уровень CD21 B-лимфоцитов и двойных отрицательных Т-клеток, что указывает на склонность к иммунной дисрегуляции [22].
Выявление пациентов с вариантами в гене DOCK11 позволило бы расширить фенотипический спектр заболевания. Т-клетки с дефицитом DOCK11 имеют сниженную трансэндотелиальную миграцию in vitro, но также увеличенную скорость миграции в условиях ограничения in vitro и in vivo. Данное обстоятельство показывает, что DOCK11 участвует в лейкоцитарном диапедезе и интерстициальной миграции. Аутовоспаление, которое наблюдалось при нескольких дефицитах, связанных с актином, ассоциировано с повышенной секрецией интерлейкина-1β и интерлейкина-18. Однако авторы не нашли доказательств такого механизма в моноцитах с дефицитом DOCK11. Вместо этого они наблюдали измененные уровни провоспалительных цитокинов и повышенный уровень ядерного фактора активированных Т-клеток (англ. nuclear factor of activated T cells 1, NFATc1) у двух пациентов. Аналогичным образом, Т-клетки от мышей с нокаутом DOCK11 показали аберрантное фосфорилирование c-Jun-N-терминальных киназ, ядерную транслокацию NFATc1 и измененную продукцию цитокинов. Эти данные указывают на участие DOCK11–CDC42 в пролиферации, активации и продукции цитокинов Т-клеток [22].
Дефекты в DOCK8, другом CDC42-активирующем GEF, вызывают сдвиг цитокинов 2-го типа и снижение пролиферации. Учитывая специфическую для стадии развития роль CDC42 в регуляции активации Т-клеток и связь между определенными пространственно-временными паттернами CDC42 и функциями Т-клеток, авторы предположили, что DOCK8 и DOCK11 регулируют зависимые от CDC42 процессы способом, зависящим от типа клеток, локализации и стимула. Отличительная клиническая картина у пациентов с дефицитом DOCK11, в отличие от дефицитов DOCK2 и DOCK8, дополнительно подтверждает идею о том, что белки семейства DOCK способствуют активации Rho-ГТФаз [22].
Терапия сложных нарушений иммунной дисрегуляции остается сложной задачей из-за необходимости применения иммунодепрессантов, введение которых еще больше увеличивает риск тяжелых и опасных для жизни инфекций. Высокая смертность среди пациентов с дефицитом DOCK11 подчеркивает важность разработки более конкретных стратегий лечения. Таким образом, дефицит DOCK11 – это новая Х-сцепленная иммуноопосредованная актинопатия, приводящая к нарушению активности CDC42 и активации STAT5, которая связана с аномальным ремоделированием актинового цитоскелета, а также с аномальным фенотипом регуляторных Т-клеток, что приводит к нарушению иммунной регуляции и тяжелой аутоиммунной патологии с ранним началом [7]. Вместе с тем остаются серьезные нерешенные вопросы, которые должны стать предметом будущих исследований.
В работе описана новая Х-сцепленная иммуноопосредованная актинопатия у мальчика с анти-NMDA-энцефалитом и гемизиготным вариантом в гене DOCK11. Наше клиническое наблюдение показывает роль гена DOCK11 в развитии тяжелого аутовоспалительного заболевания и представляет собой новый пример участия актинового цитоскелета в иммунных расстройствах человека.
С целью поиска причины заболевания, постановки молекулярного диагноза, а также принятия решения о тактике введения таких пациентов необходимо проведение генетического тестирования методом полноэкзомного или полногеномного секвенирования. Учитывая, что дефицит DOCK11 влияет на форму клеток, механическую миграцию или адгезию циркулирующих клеток, T-регуляторов и возможных иммунных клеток, находящихся в тканях, аллогенная трансплантация гемопоэтических стволовых клеток может рассматриваться как один из терапевтических методов терапии данной патологии.
1. Messacar K., Fischer M., Dominguez S.R., et al. Encephalitis in US Children. Infect Dis Clin North Am. 2018; 32 (1): 145–62. https://doi.org/10.1016/j.idc.2017.10.007.
2. Glaser C.A., Gilliam S., Schnurr D., et al. In search of encephalitis etiologies: diagnostic challenges in the California Encephalitis Project, 1998–2000. Clin Infect Dis. 2003; 36 (6): 731–42. https://doi.org/10.1086/367841.
3. Venkatesan A., Tunkel A.R., Bloch K.C., et al. Case definitions, diagnostic algorithms, and priorities in encephalitis: consensus statement of the international encephalitis consortium. Clin Infect Dis. 2013; 57 (8): 1114–28. https://doi.org/10.1093/cid/cit458.
4. Blincoe A., Heeg M., Campbell P.K., et al. Neuroinflammatory disease as an isolated manifestation of hemophagocytic lymphohistiocytosis. J Clin Immunol. 2020; 40 (6): 901–16. https://doi.org/10.1007/s10875-020-00814-6.
5. Ramirez G.A., Lanzani C., Bozzolo E.P., et al. TRPC6 gene variants and neuropsychiatric lupus. J Neuroimmunol. 2015; 288: 21–4. https://doi.org/10.1016/j.jneuroim.2015.08.015.
6. Guo Y., Audry M., Ciancanelli M., et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med. 2011; 208 (10): 2083–98. https://doi.org/10.1084/jem.20101568.
7. Boussard C., Delage L., Gajardo T., et al. DOCK11 deficiency in patients with X-linked actinopathy and autoimmunity. Blood. 2023; 141 (22): 2713–26. https://doi.org/10.1182/blood.2022018486.
8. Nosadini M., Thomas T., Eyre M., et al. International consensus recommendations for the treatment of pediatric NMDAR antibody encephalitis. Neurol Neuroimmunol Neuroinflamm. 2021; 8 (5): e1052. https://doi.org/10.1212/NXI.0000000000001052.
9. Malik D., Simon D.W., Thakkar K., et al. Genetic variation in genes of inborn errors of immunity in children with unexplained encephalitis. Genes Immun. 2022; 23 (7): 235–9. https://doi.org/10.1038/s41435-022-00185-5.
10. Tangye S.G., Al-Herz W., Bousfiha A., et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022; 42 (7): 1473–507. https://doi.org/10.1007/s10875-022-01289-3.
11. Vitaliani R., Mason W., Ances B., et al. Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol. 2005; 58 (4): 594–604. https://doi.org/10.1002/ana.20614.
12. Titulaer M.J., McCracken L., Gabilondo I., et al. Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study. Lancet Neurol. 2013; 12 (2): 157–65. https://doi.org/10.1016/s1474-4422(12)70310-1.
13. Dalmau J. NMDA receptor encephalitis and other antibody-mediated disorders of the synapse: the 2016 cotzias lecture. Neurology. 2016; 87 (23): 2471–82. https://doi.org/10.1212/wnl.0000000000003414.
14. Dalmau J., Armangué T., Planagumà J., et al. An update on anti-NMDA receptor encephalitis for neurologists and psychiatrists: mechanisms and models. Lancet Neurol. 2019; 18 (11): 1045–57. https://doi.org/10.1016/S1474-4422(19)30244-3.
15. Janssen E., Geha R.S. Primary immunodeficiencies caused by mutations in actin regulatory proteins. Immunol Rev. 2019; 287 (1): 121–34. https://doi.org/10.1111/imr.12716.
16. Ramaekers F.C., Bosman F.T. The cytoskeleton and disease. J Pathol. 2004; 204 (4): 351–4. https://doi.org/10.1002/path.1665.
17. Kamnev A., Lacouture C., Fusaro M., Dupré L. Molecular tuning of actin dynamics in leukocyte migration as revealed by immunerelated actinopathies. Front Immunol. 2021; 12: 750537. https://doi.org/10.3389/fimmu.2021.750537.
18. El Masri R., Delon J. RHO GTPases: from new partners to complex immune syndromes. Nat Rev Immunol. 2021; 21 (8): 499–513. https://doi.org/10.1038/s41577-021-00500-7.
19. Lougaris V., Baronio M., Gazzurelli L., et al. RAC2 and primary human immune deficiencies. J Leukoc Biol. 2020; 108 (2): 687–96. https://doi.org/10.1002/JLB.5MR0520-194RR.
20. Su H.C., Orange J.S. The growing spectrum of human diseases caused by inherited CDC42 mutations. J Clin Immunol. 2020; 40 (4): 551–3. https://doi.org/10.1007/s10875-020-00785-8.
21. Chen Y., Chen Y., Yin W., et al. The regulation of DOCK family proteins on T and B cells. J Leukoc Biol. 2021; 109 (2): 383–94. https://doi.org/10.1002/JLB.1MR0520-221RR.
22. Block J., Rashkova C., Castanon I., et al. Systemic inflammation and normocytic anemia in DOCK11 deficiency. N Engl J Med. 2023; 389 (6): 527–39. https://doi.org/10.1056/NEJMoa2210054.
Кожанова Татьяна Викторовна, к.м.н., доцент
ул. Авиаторов, д. 38, Москва 119620;
ул. Островитянова, д. 1, стр. 6, Москва 117513
Жилина Светлана Сергеевна, к.м.н., доцент
ул. Авиаторов, д. 38, Москва 119620;
ул. Островитянова, д. 1, стр. 6, Москва 117513
Мещерякова Татьяна Ивановна, к.м.н., доцент
ул. Авиаторов, д. 38, Москва 119620;
ул. Островитянова, д. 1, стр. 6, Москва 117513
Абрамов Александр Андреевич
ул. Авиаторов, д. 38, Москва 119620
Абидова Майа Магомедовна
ул. Авиаторов, д. 38, Москва 119620
Каминская Татьяна Святославовна, к.м.н.
ул. Авиаторов, д. 38, Москва 119620
Крапивкин Алексей Игоревич, д.м.н.
ул. Авиаторов, д. 38, Москва 119620
Заваденко Николай Николаевич, д.м.н., проф.
ул. Островитянова, д. 1, стр. 6, Москва 117513
Кожанова Т.В., Жилина С.С., Мещерякова Т.И., Абрамов А.А., Абидова М.М., Каминская Т.С., Крапивкин А.И., Заваденко Н.Н. Новая Х-сцепленная иммуноопосредованная актинопатия у мальчика с анти-NMDA-рецепторным энцефалитом и вариантом в гене DOCK11. Эпилепсия и пароксизмальные состояния. 2025;17(2):170-181. https://doi.org/10.17749/2077-8333/epi.par.con.2025.230
Kozhanova T.V., Zhylina S.S., Meshcheryakova T.I., Abramov A.A., Abidova M.M., Kaminskaya T.S., Krapivkin A.I., Zavadenko N.N. A novel X-linked immune-mediated actinopathy in a boy with anti-NMDA receptor encephalitis and variant in DOCK11 gene. Epilepsy and paroxysmal conditions. 2025;17(2):170-181. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2025.230

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































