



Содержание
Перейти к:
https://doi.org/10.17749/2077-8333/epi.par.con.2025.221
Перейти к:
Энцефалопатия развития и эпилептическая энцефалопатия (англ. developmental and epileptic encephalopathy, DEE), вызванная мутацией в гене KCNT1 (KCNT1-DEE), зарегистрирована в каталоге Online Mendelian Inheritance in Man (OMIM) под кодовым номером 614959. Альтернативные названия: DEE 14-го типа, ранняя инфантильная эпилептическая энцефалопатия 14-го типа. KCNT1-DEE чаще всего проявляется синдромом «эпилепсия младенчества с мигрирующими фокальными приступами» (англ. epilepsy in infancy with migrating focal seizures). Однако течение эпилепсий, вызванных мутациями в гене KCNT1, отличается значительным клиническим полиморфизмом. В статье описан случай тяжелого течения KCNT1-DEE у ребенка. В клинической картине доминировали фармакорезистентные фокальные приступы, глубокая умственная отсталость и спастический тетрапарез, а также микроцефалия и микросомия.
Малов А.Г., Брохин Л.Ю., Веселкова А.В. Тяжелое течение энцефалопатии развития и эпилептической энцефалопатии, связанной с KSNT1. Эпилепсия и пароксизмальные состояния. 2025;17(2):182-188. https://doi.org/10.17749/2077-8333/epi.par.con.2025.221
Malov A.G., Brokhin L.Yu., Veselkova A.V. Severe KCNT1-related developmental and epileptic encephalopathy. Epilepsy and paroxysmal conditions. 2025;17(2):182-188. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2025.221
Энцефалопатия развития и эпилептическая энцефалопатия (англ. developmental and epileptic encephalopathy, DEE), вызванная мутацией в гене KCNT1 (KCNT1-DEE), зарегистрирована в каталоге Online Mendelian Inheritance in Man (OMIM) под кодовым номером 6149591. Альтернативные названия: DEE 14-го типа, ранняя инфантильная эпилептическая энцефалопатия 14-го типа.
Ген KCNT1 (англ. potassium channel, subfamily T, member 1) расположен в локусе 9q34.3 и кодирует натрий-активируемые калиевые каналы (OMIM: 608167). Заболевание наследуется по аутосомно-доминантному типу и, как правило, возникает вследствие миссенс-мутаций de novo. Клинически KCNT1-DEE чаще всего проявляется синдромом «эпилепсия младенчества с мигрирующими фокальными приступами» (ЭМ МФП) (англ. epilepsy in infancy with migrating focal seizures). Однако мутации в гене KCNT1 могут приводить и к развитию варианта сонной гипермоторной (гиперкинетической) эпилепсии (СГЭ) (англ. sleep-related hypermotor (hyperkinetic) epilepsy), обозначенного в OMIM как “Epilepsy nocturnal frontal lobe, 5” (OMIM: 615005), что является примером фенотипической аллельной гетерогенности. При этом одинаковые мутации могут приводить как к ЭМ МФП, так и к СГЭ, и это позволяет предположить, что связь генотипа и фенотипа для мутаций KCNT1 не является прямой [1].
Наиболее полный обзор фенотипического и генотипического спектров эпилептических расстройств, связанных с мутацией в гене KCNT1, представлен в статье C.M. Bonardi et al. [2], в которой суммирован многолетний опыт исследователей из разных стран. В результате анализа клинической картины 248 человек были выявлены четыре фенотипические группы. Первую группу составили 152 пациента с ЭМ МФП, у которых наблюдались полиморфные приступы, в т.ч. мигрирующие моторные фокальные. Во вторую группу вошли 37 больных с DEE, отличными от ЭМ МФП, в т.ч. с синдромами Отахары и Веста. В третью группу включены 53 пациента с СГЭ, часто фармакорезистентной. В четвертой группе 6 человек имели другие фенотипы, в т.ч. височную эпилепсию.
Приводим клинический случай ребенка с тяжелым течением KSNT1-DEE, наблюдавшегося в течение 6 лет. Ранее девочка уже была кратко упомянута в статье 2019 г. под инициалами М.У. [3].
Первые дни жизни
Девочка родилась от вторых срочных родов путем планового кесарева сечения (рубец на матке после первых родов) с оценкой по шкале Апгар 8/9 баллов и массой тела 3690 г. На второй день жизни через 5 мин после внутримышечной инъекции (вакцинация от гепатита В) у ребенка впервые возникла серия коротких (до 10–15 с) вегетативных пароксизмов в виде апноэ и цианоза (сначала – только лица, через 10 мин – лица и туловища, а затем – общего цианоза).
На следующий день начались короткие моторные мультифокальные приступы в виде миоклоний в лице и конечностях, которые были расценены как «тремор». В выписке при переводе в возрасте 4 дней в отделение патологии новорожденных (ОПН) с диагнозом «церебральная ишемия» указано, что «со слов мамы, у ребенка отмечались судорожные подергивания конечностей», но термин «неонатальные судороги» в диагноз не внесен.
В ОПН к ежедневным мультифокальным миоклониям присоединились короткие (до 1 мин) полиморфные моторные фокальные приступы («эпилептический нистагм», тоническая адверсия глаз и головы, клонические судороги в конечностях и т.д.) с изменением латерализации как в одном приступе (миграция), так и в разных (альтернация). Нередко моторные феномены переходили из одного в другой, образуя картину «последовательных» припадков. Назначение леветирацетама (ЛЕВ) в дозе 14 мг/кг/сут привело к прекращению приступов на неделю.
На электроэнцефалограмме (ЭЭГ), зарегистрированной в это время, эпилептиформной активности (ЭА) не выявлено. При возобновлении приступов увеличение дозы ЛЕВ до 21 мг/кг/сут эффекта не принесло. На ЭЭГ впервые зарегистрированы диффузная ЭА и паттерны приступов в виде региональной эпилептической активности то с одной, то с другой стороны.
Второй ремиссии удалось достичь только через 10 дней, на третий день после добавления вальпроата натрия (ВП) в дозе 20 мг/кг/сут. Однако через 12 дней приступы вновь стали возникать ежедневно и, несмотря на увеличение дозы ВП до 30 мг/кг/сут (уровень в крови 75 мкг/л) и ЛЕВ до 28 мг/кг/сут, постепенно участились до многих за сутки. На ЭЭГ стала выявляться региональная ЭА во фронтотемпоральных или темпорально-париетальных зонах слева с периодическим диффузным распространением. При магнитно-резонансной томографии (МРТ) данных за ишемическое поражение головного мозга не получено, анализ спинномозговой жидкости без патологии, серологические маркеры внутриутробных инфекций не найдены. В связи с серийным течением приступов (с частотой до нескольких в час) в возрасте 1,5 мес девочка была переведена в неврологическое отделение с диагнозом «церебральная ишемия 2-й степени, эпилептический синдром».
Была предпринята попытка введения окскарбазепина (ОКС) в дозе 10 мг/кг/сут с одновременным снижением дозы ВП до 20 мг/кг/сут. Сначала отмечалось явное урежение приступов, но через 2 дня присоединились эпилептические спазмы по 5–20 за серию, до 5 серий в день. При увеличении дозы ОКС до 20 мг/кг/сут спазмы стали единичными, но «последовательные» приступы – ежечасными. На дозе ОКС 30 мг/кг/сут возник фокальный эпилептический статус с миграцией моторных феноменов, прервать который удалось только введением диазепама. В течение трех дней ОКС был отменен.
Возраст 2 мес
В возрасте 2 мес проведен многочасовой ЭЭГ-мониторинг (без видео), во время которого отмечена миграция ЭА во время приступа. Ритмичные альфа-подобные острые волны с постепенным снижением частоты и нарастанием амплитуды сначала возникали в одной гемисфере, а затем переходили на противоположную (рис. 1).
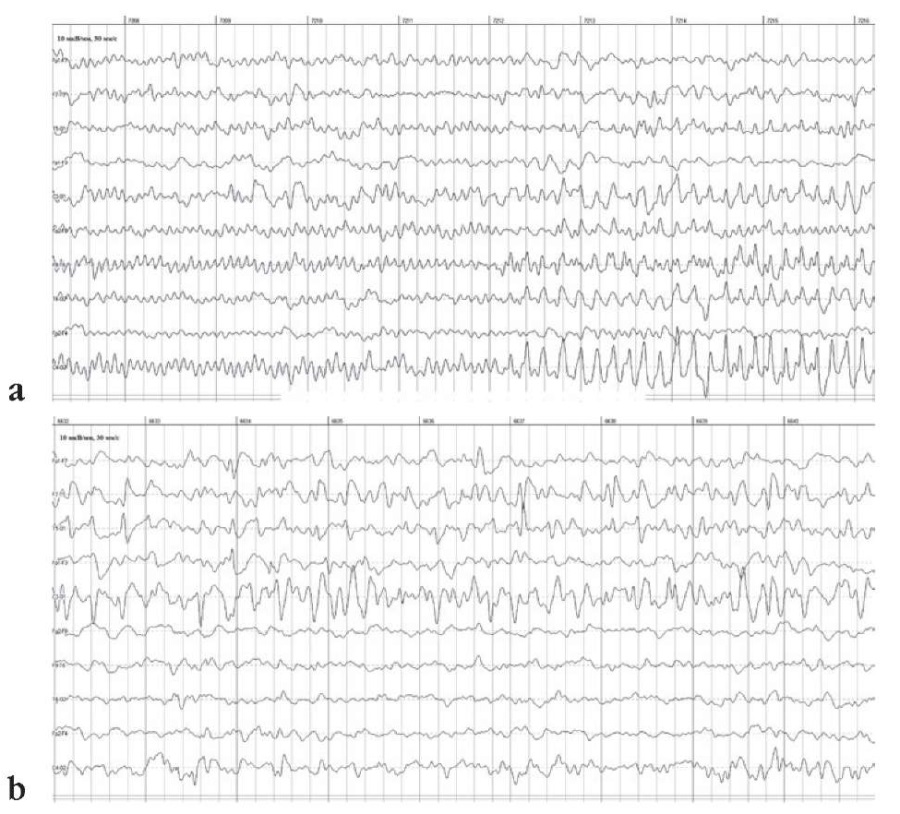
Рисунок 1. Электроэнцефалограммы (ЭЭГ) при статусном течении мигрирующих моторных фокальных приступов у пациентки М.У. (возраст 2 мес) с диагнозом «энцефалопатия развития и эпилептическая энцефалопатия, вызванная мутацией в гене KCNT1»:
а – иктальный ЭЭГ-паттерн в виде ритмичных высокоамплитудных острых волн (регулярной пилообразной активности) в отведениях от правого полушария с акцентом в С4–О2; b – миграция эпилептической активности в левое полушарие с акцентом в С3–О1
Figure 1. Patient M.U. (2 months old). Diagnosis: KCNT1-related developmental and epileptic encephalopathy. Electroencephalograms (EEG) during the status course of migrating motor focal seizures:
a – ictal EEG pattern presented as rhythmic high-amplitude sharp waves (regular sawtooth activity) in the right hemisphere, specifically in C4–O2 leads; b – migration of epileptic activity to the left hemisphere, specifically in C3–O1 leads
После этого третьим антиконвульсантом постепенно добавлен фенобарбитал (ФБ) в максимальной дозе 14 мг/кг/сут. Попытка отмены ВП привела к учащению приступов вплоть до эпилептического статуса альтернирующих гемиконвульсий, купированного диазепамом. Была диагностирована ЭМ МФП.
Возраст 2,5 мес
В 2,5 мес четвертым препаратом добавлен топирамат (ТПМ) с постепенным повышением до 4,5 мг/кг/сут, после чего без учащения приступов удалось отменить ВП, а затем ЛЕВ. Однако дальнейшее увеличение дозы ТПМ привело к учащению приступов до 70 в сутки, в связи с чем его доза была уменьшена до 4 мг/кг/сут, а позднее и до 2 мг/кг/сут. Амбулаторно на фоне дуотерапии (ФБ и ТПМ) короткие, но ежедневные приступы сохранялись с частотой от 5 до 50 раз в сутки. Отмечалась четкая зависимость от цикла «сон – бодрствование». Если в ночном сне приступы возникали 2–3 раза в час, то в дневном сне – 1 раз в час, а во время бодрствования – 1 раз в 3–6 ч. Попытки ввести клоназепам и руфинамид приводили к учащению приступов.
Возраст 3–6 мес
Кроме антиконвульсантной терапии в возрасте 3, 4 и 5 мес проводилось лечение глюкокортикостероидами в виде курсов дексаметазона (4 мг/сут № 10) с временным эффектом в виде урежения (но не прекращения) приступов. В возрасте 6 мес назначен курс метилпреднизолона 30 мг/кг/сут № 4 внутривенно капельно с последующим его пероральным приемом 2 мг/кг/сут в течение 1 мес. На этом фоне приступы временно стали более редкими (5–20 раз в сутки) и короткими (несколько секунд). Пробный прием биотина и тиамина эффекта не принес.
Возраст 9 мес
В возрасте 9 мес пациентка проходила обследование в психоневрологическом отделении Научно-исследовательского клинического института педиатрии им. академика Ю.Е. Вельтищева (обособленное структурное подразделение ФГАОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России). В этот период на фоне приема ФБ 6,8 мг/кг/сут и ТПМ 1,8 мг/кг/сут короткие моторные фокальные приступы возникали по 10–20 раз в сутки. Попытка добавить перампанел в течение четырех дней привела к учащению приступов.
На МРТ выявлено умеренное расширение желудочков мозга и наружных ликворных пространств на уровне височных долей, а также истончение мозолистого тела. После суточного электрокардиографического мониторирования диагностирована дисфункция синусового узла в виде выраженной синусовой аритмии, а также неполная блокада правой ножки пучка Гиса.
Противопоказаний к назначению антиаритмического препарата класса 1а хинидина не выявлено. Учитывая, что в литературе таргетным препаратом для терапии KSNT1-DEE считается именно хинидин [4], он был назначен в начальной дозе 15 мг/кг/сут с рекомендацией дальнейшего увеличения на 15 мг/кг/сут за месяц при отсутствии противопоказаний. Однако прием хинидина значимого эффекта не оказал, и через несколько месяцев он был отменен. Пробный прием каннабидиола к улучшению также не привел. Кетогенная диета не применялась в связи со сложностями ее проведения.
Последующие годы
В последующие годы на фоне дуотерапии (ФБ и ТПМ) короткие (до 1 мин) моторные (в основном тонические) фокальные приступы продолжались с вариабельной частотой (от 20 в сутки до 3 в неделю). Была характерна метеозависимость (в т.ч. урежение летом) и фебрильная провокация приступов (учащение при инфекциях).
Задержка психомоторного развития в виде плохого зрительного сосредоточения на фоне диффузной мышечной гипотонии отмечена через 2 нед после дебюта приступов. С 3-месячного возраста стал заметен «откат» в развитии: ребенок перестал даже кратковременно фиксировать взгляд, держать головку. Девочка никогда не поворачивалась даже на бочок, не пыталась сесть, не тянулась к предметам. Психоречевого развития практически не было: никогда не реагировала на слова, гуление отсутствовало. Когнитивные расстройства усугублялись снижением уровня бодрствования: ребенок был сонлив целый день, ненадолго просыпался только по вечерам.
Все годы девочка получает питание через соску, акт жевания не сформировался, но глотание не нарушено. Хотя оживление сухожильных рефлексов (периодически до клонуса стоп) и рефлекс Бабинского с двух сторон отмечены уже в 4 мес жизни, повышение мышечного тонуса по спастическому типу стало заметным к полугоду. Произвольных манипуляторных движений не было никогда. После 1 года сформировался центральный тетрапарез, который постепенно привел к формированию вынужденной позы в положении на спине: руки согнуты в локтевых и лучезапястных суставах и слегка пронированы, ноги согнуты в коленных и голеностопных суставах (рис. 2). Однако непроизвольные движения (например, при болях в животе) в ограниченном объеме возможны до сих пор.
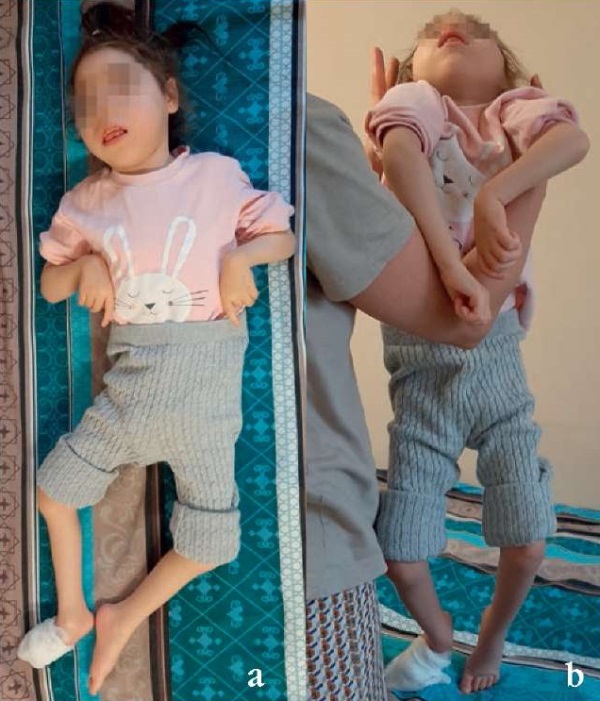
Рисунок 2. Фенотипические особенности и неврологический статус пациентки М.У. (6 лет) с диагнозом «энцефалопатия развития и эпилептическая энцефалопатия, вызванная мутацией в гене KCNT1»:
а – положение на спине; b – положение «вертикального подвешивания»
Figure 2. Patient M.U. (6 years old). Diagnosis: KCNT1-related developmental and epileptic encephalopathy. Phenotypic features and neurological status:
a – supine position; b – “vertical suspension” position
Рост мозговой части черепа стал замедляться после 1 года. К 6 годам окружность головы была 44 см (< –3 SD) при окружности грудной клетки 54 см, т.е. сформировалась постнатальная микроцефалия. Задержка физического развития стала заметна к 1 году жизни. Если в 9 мес масса тела девочки составляла 7,3 кг, а рост 70 см (индекс масы тела (ИМТ) 14,9), то постепенно набор веса практически остановился и к 6 годам она весила около 10 кг (с колебаниями до 0,5 кг в разные месяцы) при росте 96 см (ИМТ 10,9). Дефицит массы 3-й степени педиатры объясняли белково-энергетической недостаточностью. Кроме того, у девочки диагностирована хроническая гастроэзофагеальная рефлюксная болезнь с обострением в возрасте 2 года в виде эрозивного рефлюкс-эзофагита, осложненного гематомезисом, а также хронические запоры.
При нейровизуализации (МРТ 1,5 Тл) грубых структурных изменений в головном мозге не выявлялось. В возрасте 1 мес отмечены МР-признаки незрелости мозговых структур в виде задержки показателей миелинизации. В возрасте 9 мес описано умеренное расширение желудочков мозга и наружных ликворных пространств на уровне височных долей, истончение мозолистого тела. Однако на МРТ в возрасте 6 лет расширения желудочков мозга и субарахноидальных конвекситальных пространств не отмечено, при этом описаны фокусы слабоинтенсивного измененного МР-сигнала (гиперинтенсивные по Т2, T2-FLAIR, изоинтенсивные по Т1) паравентрикулярно в теменных долях, расцененные как перивентрикулярные зоны глиоза резидуального генеза.
До установления причины заболевания ребенок прошел множество исследований и диагностических процедур для уточнения вида предполагаемой генетической патологии. Кариотип без нарушений. Хромосомный микроматричный анализ расширенный: патогенного хромосомного дисбаланса не обнаружено. Аммиак в плазме 71,84 ммоль/л (норма 18–72 ммоль/л). Тандемная масс-спектрометрия (кровь) на спектр аминокислот и ацилкарнитинов: данных за наследственные аминоацидопатии, органические ацидурии и дефекты митохондриального бета-окисления не выявлено. Исследование пятен крови на болезни Помпе, Гоше, Ниманна–Пика – в пределах референсных значений.
Для исключения пиридоксин-зависимой эпилепсии проводилось пробное внутривенное введение пиридоксина в дозе 100 мг № 5 (однократно с мониторингом ЭЭГ) – без эффекта. Для исключения дефицита GluT1 определен уровень глюкозы в ликворе (2,6 ммоль/л при норме 2,2–3,3 ммоль/л) и крови (4,1 ммоль/л при норме 3,3–5,5 ммоль/л), т.е. соотношение глюкоза ликвора / глюкоза крови 0,63 (норма 0,54–0,56).
После 3 мес жизни получен результат молекулярно-генетического исследования по панели «Наследственные эпилепсии». Обнаружена ранее не описанная гетерозиготная мутация в 15-м экзоне гена KCNT1 (chr9:138660712A>G), приводящая к аминокислотной замене в 480-й позиции белка (Asp480Gly, NM_020822.2). Кроме того, выявлена ранее не описанная гетерозиготная мутация в 3-м экзоне гена SLC19A3 (chr2:228564048T>C, rs770685804), приводящая к замене аминокислоты в 128-й позиции белка (p.Tyr128Cys, NM_025243.3). Мутация, обнаруженная при применении технологии секвенирования нового поколения в гене KCNT1, была подтверждена у ребенка секвенированием по Сэнгеру, а у матери и отца данная нуклеотидная замена не выявлена.
Анализируя картину нашего клинического наблюдения в сравнении с данными литературы, можно отметить следующие особенности. Заболевание дебютировало в первые дни жизни с мультифокальных миоклоний (расцененных как «тремор»), что более характерно для ранней инфантильной DEE (синдром Отахары) [5]. Однако появление полиморфных моторных фокальных приступов с феноменами миграции и альтернации дало возможность заподозрить ЭМ МФП. Возникновение эпилептического статуса альтернирующих гемиконвульсий и мигрирующего паттерна на иктальной ЭЭГ подтвердило диагноз.
При оценке изменения приступов под влиянием антиконвульсантов наблюдались феномены «ускользания» и «аггравации». Феномен «ускользания» проявляется тем, что сначала лечение приводит к некоторому урежению приступов, но затем возникает фармакорезистентность. Феномен «аггравации» заключается в учащении приступов и появлении их новых видов при назначении препарата или повышении дозы. Со слов мамы, «почти каждый антиконвульсант был эффективен в течение нескольких дней или недель, но вскоре пароксизмы возвращались, а увеличение дозы приводило к обратному эффекту». В результате перебора множества антиконвульсантов ребенок остался на дуотерапии ФБ и ТПМ, что дало возможность относительно стабилизировать состояние и избежать повторения эпилептических статусов.
По данным литературы, для KCNT1-DEE характерно рефрактерное течение эпилепсии. Никто из пациентов с фенотипом ЭМ МФП не смог достичь длительной ремиссии по приступам, хотя использовался диапазон от 3 до 18 различных противоэпилептических препаратов [2]. Снижения частоты приступов удавалось добиться при применении ФБ (11/44), клобазама (7/43), карбамазепина (2/9) и антиаритмического препарата хинидина (13/47). Кетогенная диета была успешна в 23 случаях из 64. Эффективность хинидина специально оценивалась в метаанализе D. Xu et al. [4], охватившем 27 исследований. Из 80 пациентов с 33 мутациями KCNT1 у 20 человек наблюдалось снижение приступов на ≥50% благодаря терапии хинидином. Возраст, генотипы мутаций KCNT1, типы приступов и МРТ головного мозга не оказывали существенного влияния на терапевтический эффект хинидина. Интересно, что эта терапия часто оказывала разное воздействие у пациентов с одной и той же мутацией KCNT1, а результаты тестов хинидина in vitro не соответствовали тестам in vivo [4]. Важно отметить, что временная псевдоэффективность терапии с последующей фармакорезистентностью очень характерна для «истинных» генетических форм эпилепсии (моногенные эпилепсии и симптоматические эпилепсии при иных генетически детерминированных синдромах), в отличие от идиопатических (мультифакторных) эпилепсий [6].
Перманентные психоневрологические нарушения у ребенка проявились вскоре после начала приступов, что характерно для KCNT1-DEE [3]. Ведущими неврологическими синдромами были глубокая умственная отсталость и спастический тетрапарез. Когнитивные нарушения, обычно тяжелые, наблюдаются во всех случаях KCNT1-DEE. Однако двигательные расстройства представлены, как правило, мышечной гипо- (чаще) или гипертонией, редко – дистонией конечностей, а спастичность описана только у 5 из 16 детей с иной DEE (не ЭМ МФП) [2]. Главными соматическими проявлениями заболевания у нашей пациентки были микроцефальная форма черепа и микросомия. Микроцефалия отмечена у 60 из 149 пациентов с ЭМ МФП вследствие KCNT1-DEE [2]. Однако задержка физического развития (в 6 лет масса тела 10 кг при росте 96 см, ИМТ 10,9) с дефицитом массы 3-й степени для таких больных не характерна. Не исключено, что некоторую роль в этом мог сыграть длительный прием ТПМ, вызывающего снижение аппетита.
При нейровизуализации в нашем случае в разном возрасте описывали МРТ-признаки задержки миелинизации, «атрофической гидроцефалии» и «перивентрикулярные зоны глиоза», но грубых структурных изменений в мозге не выявлялось, что соответствует данным литературы. Так, в обширном исследовании из 118 пациентов с ЭМ МФП, обусловленной мутацией в гене KCNT1, МРТ-аномалии найдены только у 61 – в первую очередь, задержка миелинизации и кортикальная атрофия [2].
Обусловленность заболевания ребенка доказанной гетерозиготной мутацией в 15-м экзоне гена KCNT1 не вызывает сомнений, т.к. гетерозиготные миссенс-мутации в гене KCNT1 описаны у пациентов с DEE 14-го типа (OMIM: 614959). Выявленная гетерозиготная мутация в 3-м экзоне гена SLC19A3 не играет этиологической роли, поскольку у пациентов с болезнью базальных ганглиев, чувствительной к биотину и тиамину (OMIM: 607483) представлены только гомозиготные и компаунд-гетерозиготные мутации в гене SLC19A3. Более того, это заболевание проявляется экстрапирамидными и другими моторными расстройствами, а не эпилепсией. Пробный прием биотина и тиамина эффекта не приносил.
Самым сложным остается вопрос соотношения «генотип – фенотип». Известно, что как ЭМ МФП, так и СГЭ могут быть обусловлены одинаковыми мутациями в гене KCNT1 [1]. Согласно наиболее полному исследованию [2] в объединенной когорте из 248 человек было идентифицировано 64 различные мутации в KCNT1. Все они, за исключением одной, были миссенс-типа, обнаружено 24 рекуррентных (повторяющихся) мутаций. Выявлены определенные аминокислотные позиции, которые чаще всего ассоциировались с фенотипами DEE (как ЭМ МФП, так и отличными от нее): 23 из 29 пациентов с мутацией в гене KCNT1 в аминокислотной позиции 288 в области белковой петли, 21 из 21 человека с мутацией в позиции 428, 31 из 33 больных с мутацией в позиции 474 в домене RCK1 и 25 из 30 пациентов с мутацией в позиции 934 в домене RCK2 [1].
В исследовании 2022 г. [7] интернациональная команда авторов изучила функциональные эффекты 14 мутаций в гене KCNT1, проанализировала свойства калиевых токов белка KCNT1 и попыталась найти корреляцию между изменениями характеристик KCNT1 из-за мутаций и тяжестью неврологического расстройства. В результате показана положительная корреляция между тяжестью неврологического расстройства и вероятностью открытия канала KCNT1 при мембранном потенциале покоя. Это говорит о том, что мутации с усилением функции (англ. gain of function) KCNT1 вызывают эпилепсию, увеличивая проводимость калия в состоянии покоя и подавляя активность тормозных нейронов. Снижение активности потенциала действия в тормозных нейронах из-за чрезмерно высокой проводимости калия в состоянии покоя приводит к растормаживанию нейронных цепей, гипервозбудимости и судорогам.
Таким образом, эпилепсии, связанные с мутациями в гене KCNT1, являются наглядным примером клинического полиморфизма наследственных заболеваний: разные эпилептические синдромы с различным течением могут возникать при мутациях одного и того же гена. Наше клиническое наблюдение служит примером очень тяжелого течения неврологической патологии, обусловленной мутацией в гене KCNT1, с определенными особенностями, которые не полностью укладываются в рамки одного эпилептического синдрома. Можно надеяться, что дальнейшее изучение корреляций «генотип – фенотип» при KCNT1-ассоциированных эпилепсиях с учетом эпигенетических влияний даст возможность раскрыть причины различной тяжести заболевания при сходной генетической патологии.
1. https://omim.org/entry/614959?search=614959&highlight=614959.
1. Lim C.X., Ricos M.G., Dibbens L.M., Heron S.E. KCNT1 mutations in seizure disorders: the phenotypic spectrum and functional effects. J Med Genet. 2016; 53 (4): 217–25. https://doi.org/10.1136/jmedgenet-2015-103508.
2. Bonardi C.M., Heyne H.O., Fiannacca M., et al. KCNT1-related epilepsies and epileptic encephalopathies: phenotypic and mutational spectrum. Brain. 2021; 144 (12): 3635–50. https://doi.org/10.1093/brain/awab219.
3. Холин A.A., Заваденко Н.Н., Федонюк И.Д. и др. Ранняя младенческая эпилептическая энцефалопатия 14-го типа: три случая эпилепсии младенчества с мигрирующими фокальными приступами, обусловленными мутациями гена KCNT1. Журнал неврологии и психиатрии им. С.С. Корсакова. 2019; 119 (7-2): 74–82. https://doi.org/10.17116/jnevro201911907274.
4. Xu D., Chen S., Yang J., et al. Precision therapy with quinidine of KCNT1-related epileptic disorders: a systematic review. Br J Clin Pharmacol. 2022; 88 (12): 5096–112. https://doi.org/10.1111/bcp.15479.
5. Блинов Д.В. Эпилептические синдромы: определение и классификация Международной Противоэпилептической Лиги 2022 года. Эпилепсия и пароксизмальные состояния. 2022; 14 (2): 101–82. https://doi.org/10.17749/2077-8333/epi.par.con.2022.123.
6. Малов А.Г. Идиопатические и симптоматические формы генетической эпилепсии. Эпилепсия и пароксизмальные состояния. 2022; 14 (1): 91–5. https://doi.org/10.17749/2077-8333/epi.par.con.2022.107.
7. Rychkov G.Y., Shaukat Z., Lim C. X., et al. Functional effects of epilepsy associated KCNT1 mutations suggest pathogenesis via aberrant inhibitory neuronal activity. Int J Mol Sci. 2022; 23 (23): 15133. https://doi.org/3390/ijms232315133.
Малов Александр Германович, д.м.н., доцент
ул. Петропавловская, д. 26, Пермь 614000
Брохин Леонид Юрьевич
ул. Ленина, д. 13, Пермь 614015
Веселкова Ангелина Владимировна
Комсомольский пр-т, д. 43, Пермь 614000
Малов А.Г., Брохин Л.Ю., Веселкова А.В. Тяжелое течение энцефалопатии развития и эпилептической энцефалопатии, связанной с KSNT1. Эпилепсия и пароксизмальные состояния. 2025;17(2):182-188. https://doi.org/10.17749/2077-8333/epi.par.con.2025.221
Malov A.G., Brokhin L.Yu., Veselkova A.V. Severe KCNT1-related developmental and epileptic encephalopathy. Epilepsy and paroxysmal conditions. 2025;17(2):182-188. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2025.221

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































