



Содержание
Перейти к:
https://doi.org/10.17749/2077-8333/epi.par.con.2025.226
Перейти к:
В статье описаны три клинических случая наследственных нарушений, связанных с хромосомными мутациями: делециями хромосом 14 и 18. Эти наблюдения представляют профессиональный и научный интерес, т.к. относятся к раритетной неврологической патологии. Редкость данной аномалии, наличие осложнений, дороговизна инвазивной диагностики, вариабельность фенотипа, включая тяжелые врожденные пороки развития у детей с микроделециями, – все это приводит к недодиагностированию пациентов на этапе пренатальной диагностики, что влечет за собой трудности в подборе эффективной и безопасной терапии, потребность в медицинской и психосоциальной реабилитации детей в обществе. При возникновении рефрактерной эпилепсии с задержкой развития в младенческом возрасте, характерной для хромосомных микроделеций, следует проводить генетическое консультирование и обследование с целью поиска хромосомной патологии. Повышение осведомленности врачей о данном нарушении будет способствовать его своевременным диагностике и лечению.
Новикова Л.Б., Файзуллина Н.М., Акопян А.П., Зюльцле К.М. Неврологические проявления наследственных хромосомных заболеваний. Эпилепсия и пароксизмальные состояния. 2025;17(2):189-199. https://doi.org/10.17749/2077-8333/epi.par.con.2025.226
Novikova L.B., Faizullina N.M., Akopyan A.P., Ziultsle K.M. Neurological manifestations of hereditary chromosomal diseases. Epilepsy and paroxysmal conditions. 2025;17(2):189-199. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2025.226
Хромосомные заболевания занимают одно из ведущих мест в структуре причин формирования различных пороков развития, неврологических расстройств, новообразований, а также смертности в раннем периоде жизни [1]. Среди хромосомных заболеваний выделяют патологию, связанную с мутацией хромосом, к которой относятся делеции, дупликации, транслокации и инверсии. В случае делеции теряется часть генетической информации из-за потери удаленных участков генетической цепи. Такие мутации обозначаются как концевые делеции, при которых может сформироваться кольцевая хромосома. Делеции бывают терминальными (конечными) и интерстициальными (с двумя точками разрыва в пределах одного плеча какой-либо хромосомы) [2]. Как правило, при концевых делециях поломка возникает в дистальных сегментах плеча, тогда как при интерстициальных – в проксимальных сегментах. Крупные делеции часто летальны, и вообще среди всего обширного спектра хромосомной патологии делеции нередко наиболее патогенны по сравнению с транслокациями, инсерциями и др. Клинические симптомы при делециях зависят от размеров участков, затронутых перестройкой, а также от генов, содержащихся в них [2]. Подобная хромосомная патология является основой редких генетических заболеваний, таких как синдром кольцевой хромосомы r(14) и делеции хромосомы 18.
У людей с делецией 14q отсутствует часть генетического материала на длинном плече одной из копий хромосомы 14. Другая копия хромосомы 14 остается неповрежденной. Тяжелых проблем со здоровьем не возникнет, если часть генетического материала утрачена на коротком плече хромосомы 14, т.к. там не содержится особенно важных для жизнедеятельности генов [1][2]. У носителя такой мутации находят моносомию по концевым участкам в связи с концевыми делециями, при которых может сформироваться кольцевая хромосома. Синдром кольцевой хромосомы r(14) вызван одной аберрантной хромосомой 14, сломанные концы которой срастаются вместе, образуя кольцо [3–5]. Больные с r(14) имеют одну копию аномальной хромосомы в некоторых или во всех клетках. В большинстве случаев патология носит спорадический характер и возникает как случайное событие во время раннего эмбрионального развития или формирования мужских и женских половых клеток. Имеется единичное описание случаев наследственной передачи r(14) [6].
Первые научные данные об этом заболевании были представлены в 1971 г., и за последующий период в мире зарегистрировано менее 100 случаев синдрома r(14). Синдром недостаточно изучен и представляет медицинский и научный интерес [5–7]. Фенотип хромосомы r(14) характеризуется дисморфическими чертами лица, эпилептическими приступами с первого года жизни, умственной отсталостью, ретинопатией [5][7–12], рецидивирующими респираторными инфекциями, вызванными проблемами с иммунитетом [5]. Начало эпилепсии при r(14) обычно приходится на первый месяц жизни [13][14]. Приступы в основном носят генерализованный или фокальный характер, могут проявляться сенсорными или поведенческими нарушениями, потерей контроля над функциями тазовых органов [5].
Другое заболевание связано с делецией хромосомы 18. Имеется несколько тысяч известных случаев с различными ее делециями (моносомия 18p-, синдром 18p-, частичная моносомия 18p-, моносомия 18q-, синдром 18q-). У людей с делецией 18q есть только одна неповрежденная хромосома 18, а у другой некоторая часть отсутствует, что может отразиться на способности к обучению и физическом развитии. Большая часть клинических нарушений связана с наличием только одной копии генов (вместо обычных двух). Кроме того, на будущее развитие, потребности и достижения ребенка влияют его другие гены и личностные характеристики [15].
Около 1 из 40 тыс. младенцев рождается с делецией 18q. Девочки болеют в полтора раза чаще мальчиков. Несмотря на то что клинические проявления делеции 18q могут существенно различаться, есть и достаточное количество схожих черт, которые позволяют выделить потерю части длинного плеча хромосомы 18 в синдром, называемый синдромом 18q-. Впервые фенотип пациентов с данной патологией описал французский генетик Жан де Груши: в 1963 г. – делецию хромосомы короткого плеча (синдром де Груши 1-го типа) и в 1964 г. – делецию хромосомы длинного плеча (синдром де Груши 2-го типа) [16–18].
Синдром 18q-, кариотип 46XXdel, (18)(q21.3) представлен в базе данных Online Mendelian Inheritance in Man (OMIM) под номером 6018081. Исходя из локализации делеции выделяют ее проксимальный (18q11.2-q21.1) и дистальный (18q21.1-q23) типы [19]. Клинические проявления отличаются полиморфизмом, чаще всего характеризуются низким ростом, проблемами со стопами, мышечной гипотонией, задержкой речевого и умственного развития, черепно-лицевыми дисморфиями (плоское или вогнутое лицо, микроцефалия, уши с выступающим противозавитком, короткие глазные щели, гипертелоризм, эпикант, маленький рот, узкие слуховые проходы) [19]. Реже наблюдаются различные врожденные пороки развития сердца, почек, центральной нервной системы (ЦНС), задержка миелинизации, птоз, эпилептические приступы, а также офтальмологические нарушения (нистагм, диплопия, косоглазие, атрофия зрительного нерва), аномалия половых органов, дефицит иммуноглобулина A, гипотиреоз, кондуктивная тугоухость, гипопитуитаризм, кариес [5][20]. Тяжесть симптомов и клинические проявления зависят от размера и локализации отсутствующего участка хромосомы, в связи с чем трудно выделить четкий фенотип синдрома 18q- (он крайне вариабелен у различных пациентов). Проксимальные типы делеции протекают тяжелее дистальных [15][18].
Несмотря на существующую сложность стратификации пациентов с del(18) из-за схожести клинических признаков, выделен обобщенный, или «средний», фенотип больных с del(18p) и del(18q) [18]. По данным многих исследований, при обоих вариантах del(18) после рождения отмечается задержка физического и нервно-психического развития с первых месяцев жизни [18]. При del(18q) чаще, чем при del(18p), встречаются врожденные пороки развития внутренних органов. Специфического лечения del(18) не существует, проводится симптоматическая и заместительная терапия [15][21]. Прогноз для жизни зависит от тяжести врожденных дефектов органов и систем. Младенческая и ранняя детская смертность обусловлены грубыми пороками развития ЦНС. Если они корригируемые, то продолжительность жизни достаточно высокая и может составлять 60 лет. Вместе с тем подавляющее большинство больных с del(18) социально дезадаптированы и нуждаются в пожизненной поддержке семьи и государства [15][18].
Для иллюстрации изложенного материала приводим клинические случаи синдрома r(14) и синдрома 18q-, кариотип 46XXdel.
В психоневрологическом отделении (ПНО) № 1 Детского центра психоневрологии и эпилептологии (ДЦПНиЭ) ГБУЗ «Республиканская детская клиническая больница» г. Уфы (ГБУЗ РДКБ) наблюдались два ребенка мужского пола с синдромом r(14): пациент К. (род. 02.02.2018 г.) и пациент Х. (род. 29.03.2019 г.) .
Ведущим проявлением заболевания у обоих детей была рефрактерная эпилепсия, которая в первом случае дебютировала в возрасте 11 мес приступами судорог и нарушением сознания, а во втором случае первый судорожный приступ развился в возрасте 7 мес. Оба пациента были госпитализированы и обследованы.
Анамнез заболевания
Пациент К. – ребенок от третьей беременности (протекавшей на фоне хронической гипоксии плода, диффузного узлового зоба матери), вторых родов. Во время беременности мать принимала эутирокс по поводу диффузного узлового зоба. Роды на 38–39-й неделе путем планового кесарева сечения. При рождении масса тела ребенка 3190 г. Выписан на 8-е сутки. Ранее развитие соответствовало возрасту.
Пациент Х. – ребенок от четвертой беременности (резус отрицательный), вторых родов. Во время беременности мать перенесла острую респираторную вирусную инфекцию, были изменения в анализах: анемия, протеинурия. Роды на 38-й неделе. Масса тела ребенка 3900 г, окружность груди 34 см, оценка по шкале Апгар 7/9 баллов. Из роддома выписан на 4-е сутки. Задержка моторного развития с 6 мес, задержка речевого развития с 11 мес. Ходит самостоятельно с 1 года 6 мес. Первые слова с 1 года.
Неврологический статус
В обоих случаях имелись особенности строения лицевого черепа и скелета: у пациента К. микроцефалия, эпикант, антимонголоидный разрез глаз (опущены наружные углы глазных щелей), плоская спинка носа, вывернутые ноздри, узкое нёбо, короткая шея, воронкообразная грудь, аномалии пигментации кожи и сетчатки. Зрачки D=S, реакция на свет живая. Взор фиксирует, за предметами следит. Глотание и фонация не нарушены. Голову держит, переворачивается, сидит, встает у опоры, передвигается у опоры, ходит с поддержкой. Тонус мышц – гипотония с обеих сторон. Опора на полные стопы. Патологические знаки отсутствуют. Менингеальных знаков нет. Познавательно-ориентировочная деятельность с отставанием по возрасту. Задержка речевого развития.
У пациента Х. микрогнатия, прогнатия, высокое нёбо, оттопыренные уши, неправильный рост зубов (диастемы), удлиненные конечности. Черепно-мозговые нервы: глазные щели D=S, зрачки D=S, фотореакция живая, следит за предметами, альтернирующее косоглазие. Глотание и фонация не нарушены. Гипотония мышц, D=S. Сухожильные рефлексы живые, D=S. Рефлекс Бабинского положительный с обеих сторон. Сидит, переворачивается, ходит самостоятельно с широкой базой. Гипермобильность суставов кистей. Познавательно-ориентировочная деятельность умеренно снижена, грубая задержка речевого развития.
Инструментальные методы обследования
Электроэнцефалография (ЭЭГ) проводилась в обоих случаях неоднократно.
У пациента К. на 30-й день пребывания в стационаре на фоне противосудорожной терапии во время сна произошел генерализованный тонико-клонический приступ. На ЭЭГ во время приступа зарегистрированы редуцированные комплексы «пик – волна» амплитудой 40 мкВ по всем отведениям длительностью 6 с, заканчивающиеся волнами дельта-диапазона амплитудой до 500 мкВ с преобладанием в передних отведениях (рис. 1).
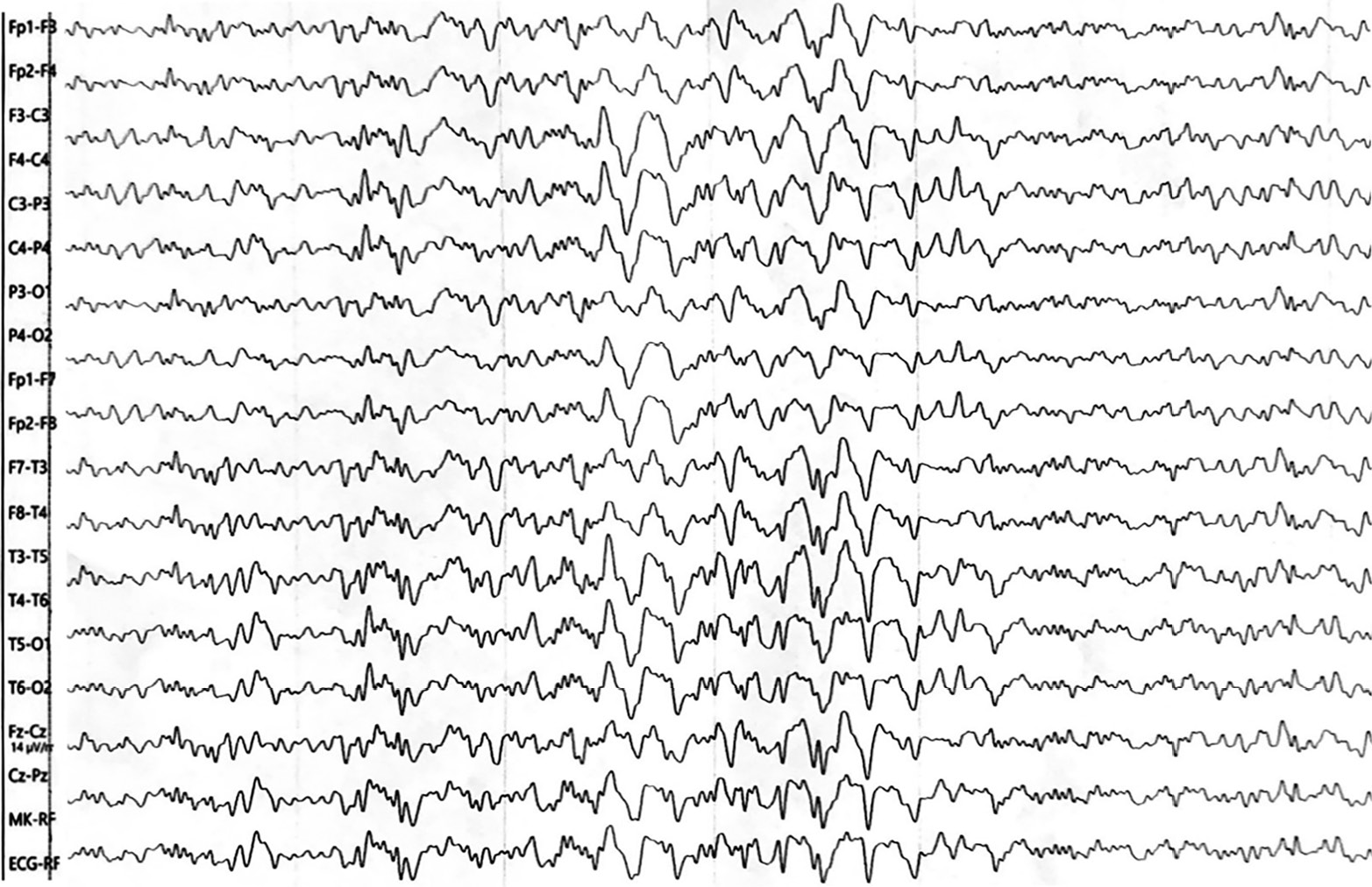
Рисунок 1. Пациент К. (возраст 11 мес). Электроэнцефалограмма в период эпиприступа
Figure 1. Patient K. (aged 11 months). Еlectroencephalogram during epileptic seizure
У пациента Х. многократно проводились записи ЭЭГ, в т.ч. ночной видео-ЭЭГ-мониторинг: во время сна через 20 дней после первого судорожного приступа, в возрасте 1 год 7 мес и в возрасте 3 года 2 мес. При видео-ЭЭГ-мониторинге в состоянии сна регистрировалась полиморфная медленноволновая активность, преобладающая по периоду и амплитуде до 350 мкВ в теменно-затылочных и височных областях, без асимметрии. Вторая стадия фазы медленного сна характеризовалась появлением билатерально-синхронных вспышек частотой 11–13 Гц, К-комплексов в виде полифазных потенциалов. Эпилептиформная активность не зарегистрирована. Во время записи ЭЭГ в возрасте 4 года 3 мес произошел приступ длительностью до 2 мин с появлением на ЭЭГ миографических артефактов в височных областях, диффузных, ритмичных, редуцированных (замедленных) комплексов, комплексов «острая – медленная волна» амплитудой до 300 мкВ в лобно-височных областях (рис. 2).
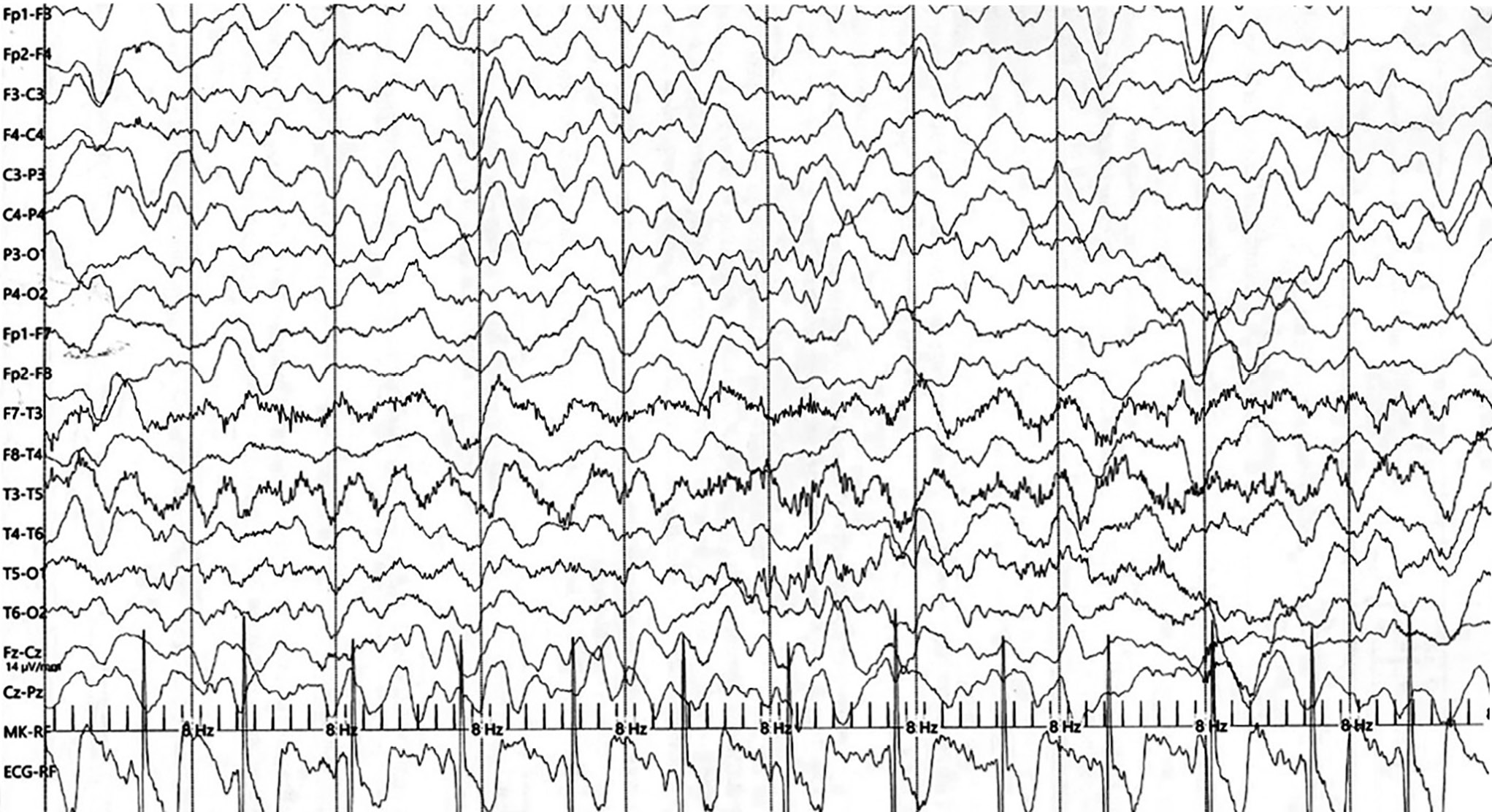
Рисунок 2. Пациент Х. (возраст 4 года 3 мес). Электроэнцефалограмма во время эпиприступа
Figure 2. Patient X. (aged 4 years 3 months). Electroencephalogram during an epileptic seizure
На магнитно-резонансной томографии (МРТ) головного мозга у пациента К. при поступлении обнаружены косвенные признаки последствия гипоксически-ишемического поражения головного мозга с проявлениями внутричерепной гипертензии, кисты кармана Ратке в виде образования жидкостной кистоподобной структуры размерами до 2×3×4 мм между аденогипофизом и нейрогипофизом (рис. 3).
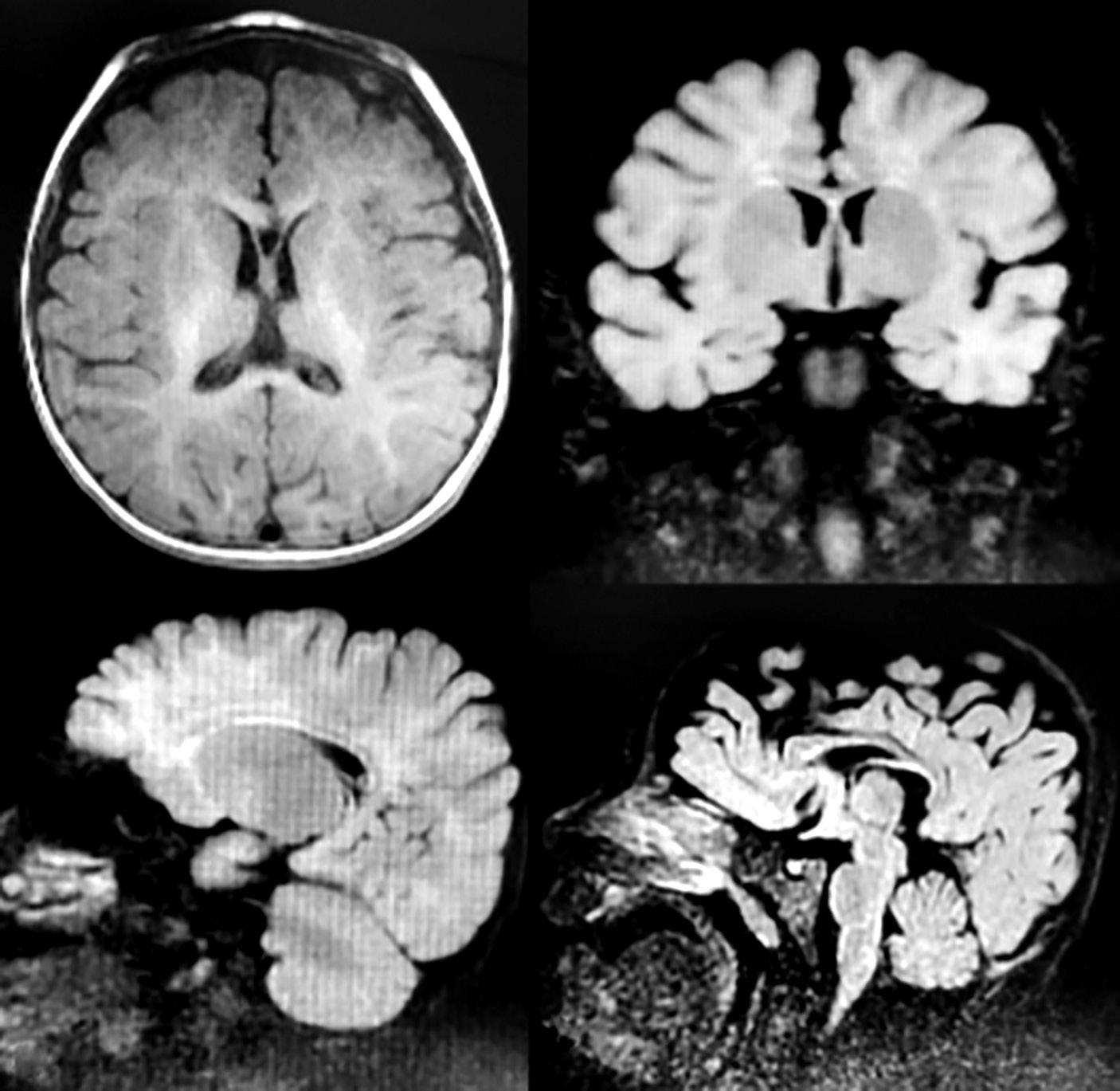
Рисунок 3. Пациент К. (возраст 11 мес). Магнитно-резонансные томограммы головного мозга:
a, b – признаки внутричерепной гипертензии; c, d – киста кармана Ратке
Figure 3. Patient K. (aged 11 months). Brain magnetic resonance images:
a, b – signs of intracranial hypertension; c, d – Rathke’s cleft cyst
На МРТ головного мозга у пациента Х. в возрасте 4 года 2 мес: все отделы мозга сформированы. Мозолистое тело неравномерно умеренно истончено. Боковые желудочки асимметричны, передние рога сужены. Перивентрикулярно в белом веществе отмечены диффузные зоны повышения МР-сигнала в режиме FLAIR. Выявлено незаращение прозрачной перегородки с образованием ликворосодержащей полости между передними рогами боковых желудочков шириной до 4 мм. Между треугольниками боковых желудочков определялась ликворная полость шириной до 8 мм. Субарахноидальные пространства конвекситальной поверхности полушарий большого мозга в лобно-теменных областях и передних отделах височных областей умеренно неравномерно расширены. Борозды лобных и теменных долей углублены. Сильвиевы щели несколько расширены. Срединные структуры не смещены. Отмечен умеренный периневральный отек по ходу зрительных нервов. Зафиксировано низкое расположение синусного стока. Большая цистерна мозга локально расширена до 27×52×20 мм (нижняя ретроцеребеллярная киста?). Заключение: «Изолированная гипоплазия нижних отделов червя мозжечка. Гипогенезия мозолистого тела. Незаращение прозрачной перегородки» (рис. 4).
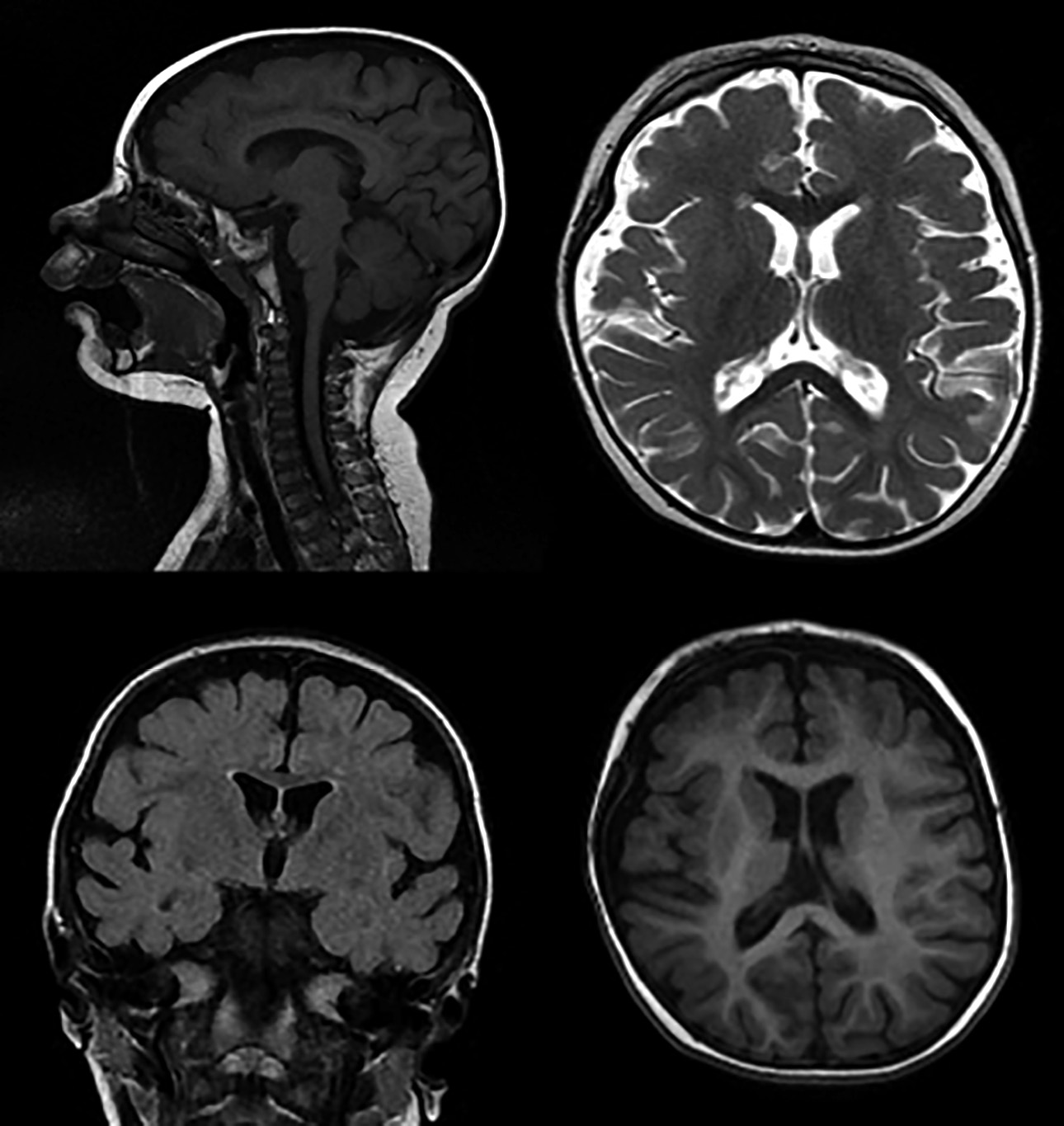
Рисунок 4. Пациент Х. (возраст 4 года 2 мес). Магнитно-резонансные томограммы головного мозга:
a – изолированная гипоплазия нижних отделов червя мозжечка, гипогенезия мозолистого тела; b–d – незаращение прозрачной перегородки
Figure 4. Patient Х. (aged 4 years 2 months). Brain magnetic resonance images:
a – isolated hypoplasia of the lower cerebellar vermis parts, corpus callosum hypogenesis; b–d – non-closure of the septum pellucidum
Результаты тандемной масс-спектрометрии на выявление наследственных болезней обмена веществ у обоих пациентов отрицательные.
Генетическое исследование
В связи с неоднозначной клинической картиной было решено провести кариотипирование в ГБУЗ «Республиканский медико-генетический центр» г. Уфы с целью выявления возможной хромосомной патологии. У обоих пациентов выявлен мужской аномальный кариотип – кольцевая хромосома 14 (46XY, r(14)(p12q32)).
Диагноз пациента К.: «Q99.8 хромосомное заболевание, кольцевая хромосома 14. Генерализованные тонико-клонические приступы, генерализованная генетическая эпилепсия. Задержка психоречевого развития, микроаномалии лицевого черепа».
Диагноз пациента Х.: «Q99.8 хромосомное заболевание, кольцевая хромосома 14. Тонические эпилептические приступы, генерализованная генетическая эпилепсия. Пирамидно-атактический синдром. Задержка психоречевого развития. Микроаномалии развития. Изолированная гипоплазия нижних отделов червя мозжечка. Гипогенезия мозолистого тела. Гипермобильность суставов кистей».
Особенности течения эпилепсии
У пациента К. в отделении наблюдалась серия эпиприступов – несколько приступов с интервалом 5 мин, в течение которых сознание относительно восстанавливалось, с периодичностью каждые 2–3 дня. Во время приступа зрачки расширены, лицо гиперемировано, клонико-тонические судороги в течение нескольких секунд. При нарушении дневного сна (чуткий, беспокойный сон) приступы учащались. У пациента Х. во время приступа наблюдались потеря сознания, генерализованные тонические судороги в конечностях, девиация глазных яблок вверх. Длительность приступа от 1 до 2 мин, частота 1–2 раза в неделю.
Одной из основных задач терапевтической тактики было купирование приступов. Пациенту К. назначалась монотерапия вальпроевой кислотой по 127 мг 3 раза в день, которая не дала ожидаемого результата. Только добавление еще двух противосудорожных препаратов (леветирацетама по 200 мг 2 раза в день и фенобарбитала по 5 мг 1 раз в сутки) дало положительный эффект.
Пациенту Х. после первых судорожных приступов в возрасте 7 мес проводилась противосудорожная терапия раствором леветирацетама и суспензией окскарбазепина без эффекта. Купирование приступов и достижение ремиссии в течение 2 лет 4 мес произошло на терапии вальпроевой кислотой в сиропе по 3,5 мл 3 раза в день и топирамата по 1 таблетке (50 мг) 2 раза в день. При рецидиве тонических приступов к проводимой терапии был добавлен фенобарбитал в дозах 6,25 мг утром и 12,5 мг вечером, что привело к ремиссии. Катамнез 8 мес на фоне противосудорожной терапии приступов не наблюдался.
Оба пациента находятся под наблюдением невролога, педиатра по месту жительства, при необходимости проводится консультация эпилептолога.
В ПНО № 1 ДЦПНиЭ ГБУЗ РДКБ в плановом порядке 13.01.2023 г. поступила пациентка Я. женского пола в возрасте 3 лет (род. 24.12.2019 г.). Со слов мамы, ребенок не разговаривает, ходит неустойчиво.
Анамнез заболевания
Ребенок от первой беременности, первых родов. Беременность у матери протекала на фоне анемии, урогенитальных инфекций, фетоплацентарной недостаточности. Наблюдалась задержка внутриутробного развития плода. Перинатальный скрининг не выявил грубых хромосомных отклонений. Роды в срок 37 нед путем кесарева сечения (раннее отхождение околоплодных вод). Девочка с массой тела 2440 г (маловесный для гестационного возраста плод), окружность головы 31 см, оценка по шкале Апгар 8/9 баллов.
С 24.12.2019 г. по 26.12.2019 г. проводилась искусственная вентиляция легких. У ребенка обнаружены врожденные пороки развития – врожденная диафрагмальная грыжа (истинная справа), гипоплазия легкого. Торакоскопическая диафрагморафия (пластика диафрагмы) справа выполнена 01.01.2020 г. Осмотр неонатолога выявил перинатальное поражение ЦНС, синдром мышечной гипотонии, синдром двигательных нарушений. Кандидоз новорожденного. Открытое овальное окно, недостаточность кровообращения 0. Из родильного дома ребенок был переведен в отделение патологии новорожденных и недоношенных детей.
Наследственность не отягощена, возраст матери 25 лет, возраст отца 40 лет, оба здоровы. Задержка развития с первого года жизни. Начала ходить самостоятельно в возрасте 2 года 6 мес неустойчиво, не разговаривает. В анамнезе частые респираторно-вирусные инфекции.
Неврологический статус
Сознание ясное. Познавательно-ориентировочная деятельность снижена. Окружность головы 45 см. Большой родничок закрыт. Черепно-лицевые дисморфии – микроцефалия, гипертелоризм, узкие глазные щели, сглаженный фильтрум, маленький рот, плоское лицо, эпикант, плоская переносица. Сглаженность завитков ушных раковин. Плосковальгусные стопы. Черепно-мозговые нервы: зрачки D=S, реакция на свет живая, взор фиксирует, взгляд за предметами прослеживает, легкая асимметрия носогубных складок, глотание и фонация не нарушены. Мышечная гипотония, D=S. Сидит, ползает, ходит с поддержкой. Сухожильные рефлексы низкие, D=S. Патологических рефлексов нет. Менингеальные знаки отрицательные.
Инструментальные методы обследования
По данным рентгенографии органов грудной клетки, легочная ткань вздута. Легочный рисунок усилен за счет сосудисто-интерстициального компонента, сгущен во внутренней легочной зоне справа. Купол диафрагмы четкий, ровный.
Электрокардиография: синусовая тахикардия (частота сердечных сокращений 115 уд/мин, электрическая ось сердца вправо. Эхокардиография: камеры сердца не увеличены, клапаны не изменены, открытое овальное окно 0,3 см, шунт лево-правый. Сократительная функция миокарда удовлетворительная.
Результаты ультразвукового исследования органов брюшной полости и почек без патологий. Нейросонография без патологии.
Выставлен диагноз: «Другие делеции части хромосомы (синдром де Груши), врожденная диафрагмальная грыжа (истинная справа). Операция от 01.01.2020 г.: торакоскопическая диафрагморафия справа. Гипоплазия и дисплазия легкого. Поражение ЦНС вследствие хромосомной патологии – синдром де Груши. Маловесный для гестационного возраста плод. Синдром мышечной гипотонии, синдром двигательных нарушений. Кандидоз новорожденного. Открытое овальное окно, недостаточность кровообращения 0».
ЭЭГ: немодулированный, нерегулярный альфа-ритм частотой 8–9 Гц, амплитудой до 60 мкВ, в центральных, теменных областях в сочетании с медленными волнами тета-диапазона амплитудой до 65 мкВ, умеренно выраженный бета-ритм частотой 20–22 Гц, амплитудой до 10 мкВ в расширенной зоне без признаков межполушарной асимметрии и специфической патологической активности (рис. 5). Проба с ритмической фотостимуляцией фотопароксизмального ответа не вызвала.
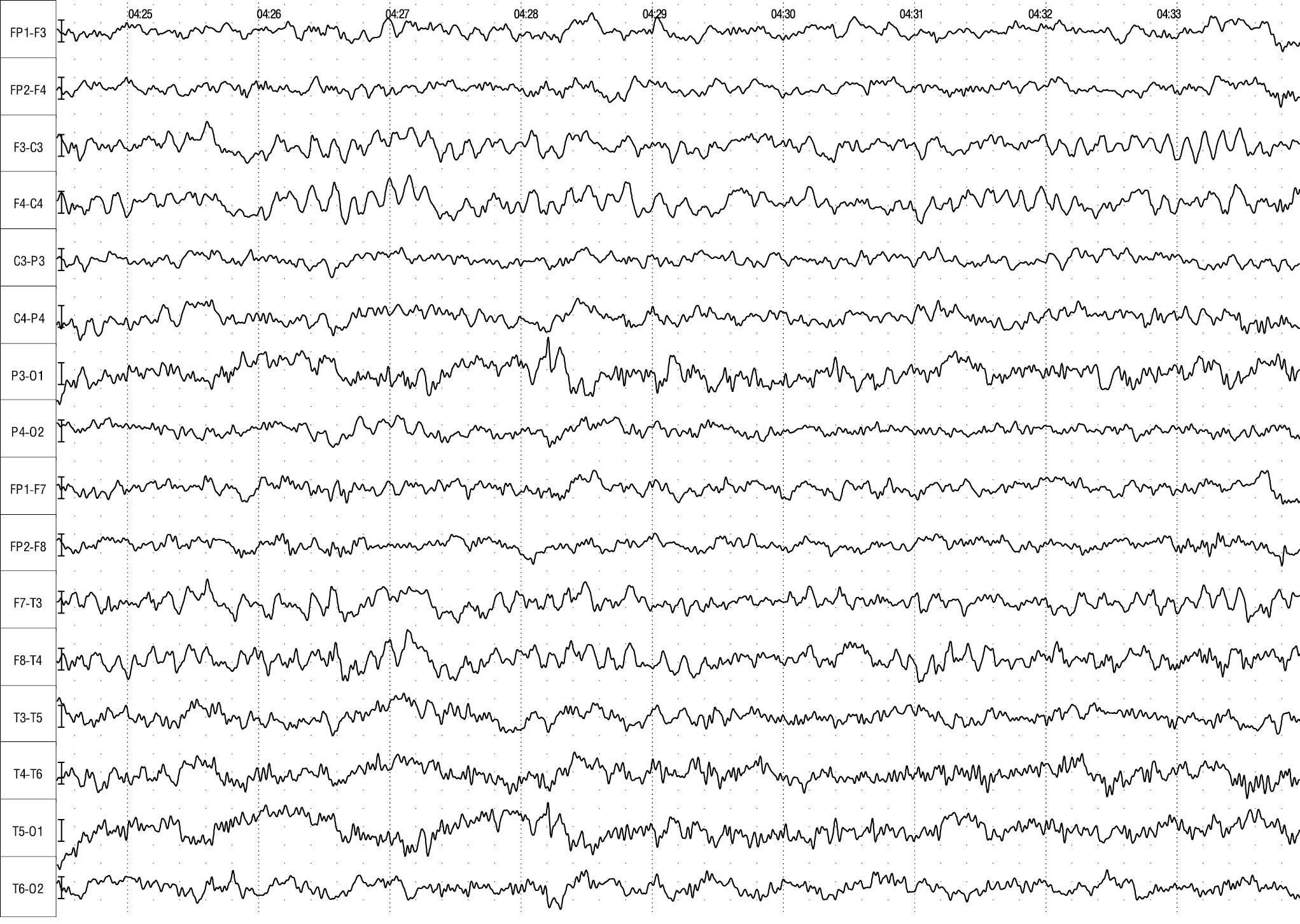
Рисунок 5. Электроэнцефалограмма пациентки Я. (возраст 3 года)
Figure 5. Рatient Ya. (aged 3 years) electroencephalogram
На МРТ головного мозга без контрастного усиления: дифференцировка серого и белого веществ сглажена. Очагов и диффузных зон патологической плотности в веществе головного мозга не выявлено. Мозолистое тело истончено. Субарахноидальные пространства сужены. Борозды не углублены. Желудочковая система обычной конфигурации. Срединные структуры не смещены. Гипофиз обычного положения и размеров. Отмечается низкое расположение синусного стока. Стволовые структуры без патологических сигналов. Миндалины мозжечка не опущены (рис. 6).
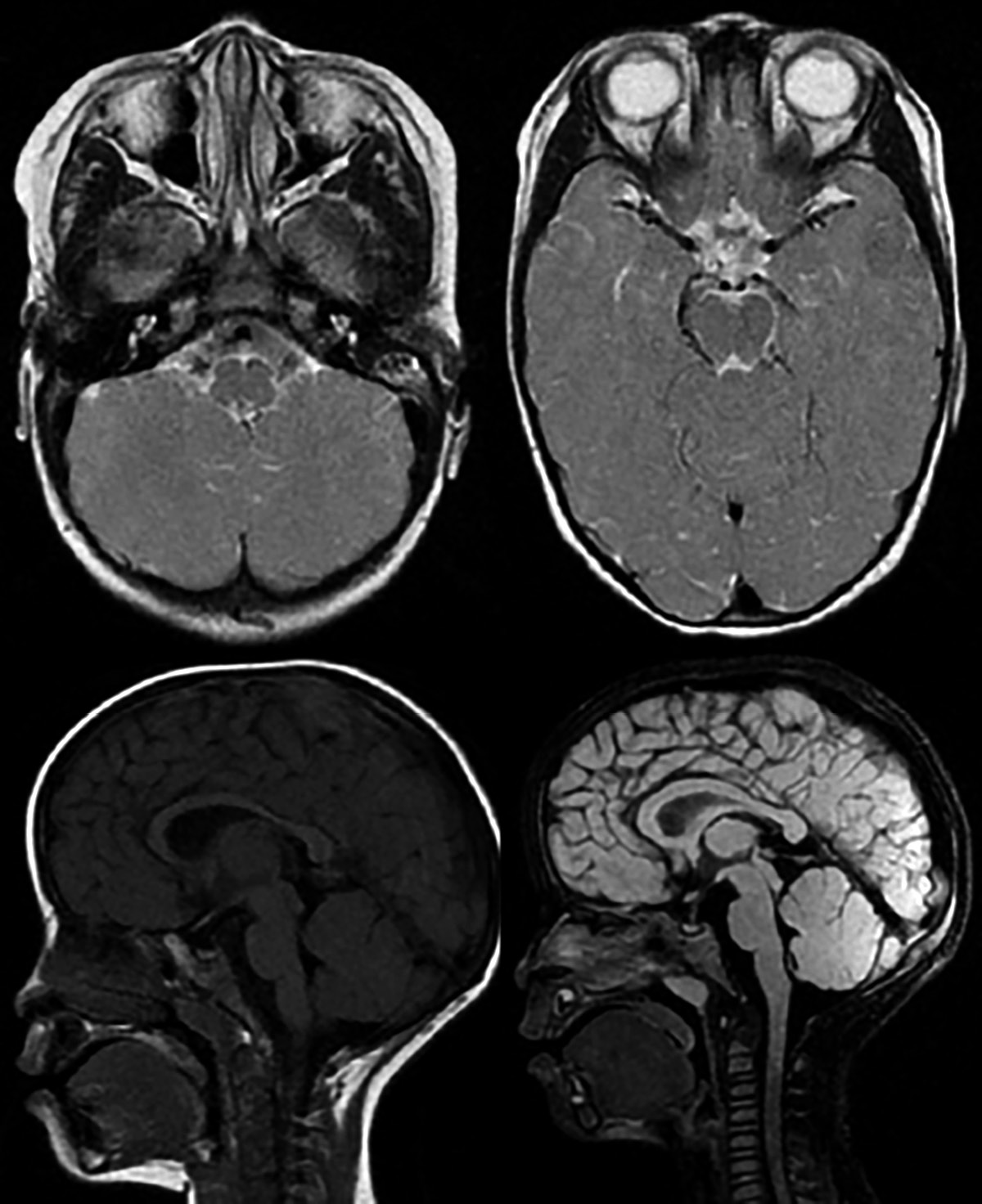
Рисунок 6. Магнитно-резонансные томограммы головного мозга пациентки Я. (возраст 3 года):
a, b – субарахноидальные пространства сужены, дифференцировка серого и белого веществ сглажены; c, d – дисгенезия мозолистого тела, низкое расположение синусного стока
Figure 6. Рatient Ya. (aged 3 years) brain magnetic resonance images:
a, b – narrowed subarachnoid spaces, smoothed differentiation of gray and white matter; c, d – corpus callosum dysgenesis, low location of the sinus drainage
На электронейромиографии нижних конечностей с регистрацией f-волн показатели проводимости по nn. peroneus, tibialis, suralis d/s без особенностей.
Генетическое исследование
Проведено кариотипирование в ГБУЗ «Республиканский медико-генетический центр» г. Уфы. Выявлена частичная моносомия хромосомы 18 (синдром 18q-), кариотип 46XXdel(18)(q21.3).
Диагноз
Пациентка консультирована генетиком, офтальмологом, логопедом, дефектологом, ортопедом. Заключение офтальмолога: OU-нистагм, частичная атрофия зрительного нерва, ангиопатия сетчатки. Заключение логопеда: задержанное речевое развитие. Заключение дефектолога: задержанное психическое развитие. Заключение ортопеда: патологии опорно-двигательного аппарата не выявлено.
Выставлен диагноз: «Q93.5. Другие делеции части хромосомы. Частичная моносомия хромосомы 18 (синдром 18q-) с врожденной диафрагмальной грыжей (оперирована), задержкой психоречевого развития, мышечной гипотонией, черепно-лицевыми дисморфизмами, ротаторным нистагмом, частичной атрофией зрительного нерва».
Терапия
Проводилась нейрометаболическая терапия, физио-, кинезио-, иглорефлексотерапия. Пациентка выписана на 8-й день (20.01.2023 г.) под наблюдение невролога, педиатра по месту жительства, даны необходимые рекомендации по дальнейшей реабилитации. Повторные стационарные сеансы терапии осуществлялись в течение 10 дней. Далее девочка неоднократно получала стационарное лечение в ПНО № 1.
Поскольку другие хромосомные нарушения могут иметь сходные признаки, для подтверждения диагноза необходимо применение современных молекулярно-цитогенетических методов исследования (кариотипирование культуры лимфоцитов, хромосомный микроматричный анализ, FISH-диагностика) [18]. Метод цитогенетического исследования обычно может идентифицировать кольцевую хромосому после того, как с помощью молекулярного кариотипирования устанавливается терминальная делеция хромосом 14q, 18q. Возможные аномалии генома часто имеют небольшие размеры, не позволяющие увидеть их при стандартном кариотипировании, но они могут значительно влиять на клиническую картину, а в сочетании с другими нарушениями – увеличить тяжесть патологических проявлений, что необходимо знать для медико-генетического консультирования семьи. Разработана пренатальная диагностика. Во многих наблюдениях делеции хромосом 14q, 18q являлись случайной находкой во время проведения хорионбиопсии, амниоцентеза у беременных женщин с установленными пороками развития плода по результатам пренатального ультразвукового скрининга [18]. В случаях отягощенного семейного анамнеза цитогенетическая экспертиза клеток ворсин хориона и/или амниотической жидкости проводится целенаправленно.
Опубликованы данные о вариантах делеции хромосомы 14 с учетом дефицита отдельных генов. Так, клинические проявления делеции 14q13 могут различаться в зависимости от того, какого она размера, а также сколько и какие именно гены были утрачены. В целом предполагается, что существует две группы людей с делецией 14q13: с легкими и тяжелыми симптомами. Делеции, затрагивающие ген NKX2-1, приводят к чрезвычайно вариабельному виду патологии, известному как синдром «мозг – легкие – щитовидная железа». Наиболее общие черты делеции хромосомы 14q13, включающей ген NKX2-1: задержка в развитии, задержка в развитии речи, различная степень трудности в обучении или неспособность к обучению, гипотиреоз, хореоатетоз, возможные нарушения в работе легких могут привести к затруднению дыхания и респираторным инфекциям. Потеря соседнего гена PAX9 может вызвать гиподонтию/олигодонтию (отсутствие зубов). Ген RALGAPA1 считается причиной неврологических расстройств, связанных с делецией хромосомы 14q. Дети, имеющие только одну копию этого гена, обладают теми же отличительными особенностями, что и дети с мутациями в нем, а именно: нарушение умственного развития, регресс речевого развития в детстве, проявления аутического поведения, задержка в развитии и гипотония. Также предполагают, что дисфункция гена RALGAPA1 может быть связана с судорожным синдромом [3][4].
Если затронуты соседние участки хромосомы, то последствия могут быть более тяжелыми. Наиболее серьезные симптомы обнаружены у людей с делециями, распространяющимися на участок 14q12, где расположен ген FOXG1, потеря которого приводит к тяжелой форме инвалидности. В случае, если затронут ген FOXG1 (или делеция происходит относительно близко к нему), обнаруживается ряд клинических особенностей, включающий малый размер головы (микроцефалия), агенезию мозолистого тела (нарушение развития сплетения нервных волокон, соединяющих правое и левое полушария), необычные черты лица, низкий мышечный тонус и судорожный синдром [3][4].
Наиболее общие черты микроделеций хромосомы 14q12-14q13.1: задержка в развитии, замедленные темпы роста головы (микроцефалия), трудности при кормлении, рефлюкс и нестабильный набор веса (задержка физического развития), запоры, низкий мышечный тонус или комбинация повышенного и пониженного мышечного тонуса, необычные черты лица, врожденные затруднения дыхания, апноэ, на МРТ нарушение развития мозолистого тела (сплетение нервных волокон, соединяющее левое и правое полушария), судорожный синдром / судорожные движения, бруксизм, холодные кисти и стопы, нарушения сна, частые инфекционные заболевания (пневмония, воспаления среднего уха), пневмония может быть вызвана рефлюксом (аспирацией), повторяющиеся (стереотипные) движения, походка может быть неустойчивой, слабое зрение, раздражительность и беспричинный плач у новорожденных. Остается вопрос о дальнейшей генетической диагностике с помощью секвенирования экзома для уточнения точковых мутаций эпилептических энцефалопатий [3][4].
В литературе есть данные о вариантах делеции хромосомы 18 с учетом дефицита отдельных генов. Особенности синдрома 18q- являются результатом потери некоторого количества разных генов. В двух исследованиях с участием более 50 человек все делеции 18q были разными, что говорит об отсутствии единой начальной точки поломки длинного плеча хромосомы 18. Следовательно, у большинства людей с делецией 18q отсутствуют разные, но часто пересекающиеся части хромосомы [15].
Исследования поврежденных участков хромосомы и признаков, за которые отвечают гены, локализованные на этих критических участках, позволяют понять патогенез нарушений в развитии больного ребенка. Например, низкорослость, задержка миелинизации, узкие (или отсутствующие) слуховые ходы и аномалия стоп у больных с синдромом 18q- были картированы в регионе 18q22.3 и 18q23. Недостаточность гипофиза при синдроме 18p- определена в регионе 18p11.32 и 18p11.21. Корреляции между генотипом и фенотипом также имеют значение в подборе персональных схем терапии и определении прогноза для конкретного больного [18].
Специфические черты для синдрома 18q-: демиелинизация и дефект синтеза основного миелинового белка ЦНС. В настоящее время не установлен ген, делеция которого вызывает нарушения миелинизации, но обсуждается роль гена – кандидата MBP (англ. myelin basic protein) в дистальном участке 18q, экспрессирующегося в олигодендроцитах [1].
В исследовании С. Zweier et al. (2008 г.) показано, что ген TCF4 в участке 18q21 отвечает за синдром Питта–Хопкинса, характеризующийся трудностями с обучением, особыми чертами лица (большой клювовидный нос, широкий рот и мясистые губы) и проблемами с дыханием [22].
Клинические проявления отличаются полиморфизмом, эпилептические приступы в рамках данной патологии могут быть генерализованными и фокальными. Эпилепсия при хромосомной патологии по этиопатогенезу относится к наследственным эпилепсиям и, как показывают данные литературы и наши наблюдения, отличается фармакорезистентностью [2][23–26]. Поэтому необходимо начинать противоэпилептическую терапию препаратами вальпроевой кислоты как средством первой линии, однако монотерапия такими препаратами не была эффективной. Фенобарбитал оказался средством достижения ремиссии в сочетании с проводимой противосудорожной терапией. Исследования синдрома 18q- демонстрируют, что эта хромосомная аномалия клинически связана, прежде всего, с возможной аномалией мозга и различными формами умственной отсталости у детей, а также с врожденными пороками и нарушениями развития [1][15][18].
Редкость данной патологии, наличие осложнений, дороговизна инвазивной диагностики, вариабельность фенотипа, включая тяжелые врожденные пороки развития у детей с микроделециями, – все это приводит к недодиагностированию пациентов на этапе пренатальной диагностики с последующей тяжестью лечения, медицинской и психосоциальной реабилитации детей в обществе. При возникновении рефрактерной эпилепсии в младенчестве с задержкой развития по неизвестной причине следует провести молекулярно-цитогенетическое обследование: хромосомный микроматричный анализ, панели генов эпилепсии (секвенирование нового поколения, секвенирование всего экзома) и медико-генетическое консультирование.
1. https://omim.org/entry/601808#de1964.
1. Камалтынова Е.М., Салюкова О.А., Федорова О.С. и др. Случай делеции длинного плеча хромосомы 18 у ребенка 2 месяцев. Педиатрия. Журнал имени Г.Н. Сперанского. 2006; 85 (6): 120–2.
2. Кравец В.С., Юров Ю.Б., Данцев И.С. и др. Редкая интерстициальная делеция хромосомы 11: необходимость молекулярноцитогенетической диагностики. Российский вестник перинатологии и педиатрии. 2016; 61 (4): 196–7.
3. Unique. Understanding Rare Chromosome and Gene Disorders. Делеции 14q12. URL: https://rarechromo.org/media/translations/Russian/14q12%20deletions%20Russian%20FTNW.pdf (дата обращения 05.09.2024).
4. Unique. Understanding Rare Chromosome and Gene Disorders. Синдром делеции 14q13. URL: https://www.rarechromo.org/media/translations/Russian/14q13%20Deletions%20Russian%20FTNW.pdf (дата обращения 05.09.2024).
5. Ivanoff A.E., Ivanoff C.S. Ring chromosome 14 syndrome: what the dentist should know to manage children with r(14) effectively. Folia Med. 2023; 65 (1): 20–9. https://doi.org/10.3897/folmed.65.e71784.
6. Новикова Л.Б., Акопян А.П., Латыпова Р.Ф., Файзуллина Н.М. Случаи фармакорезистентной эпилепсии при хромосомной патологии. Эпилепсия и пароксизмальные состояния. 2024; 16 (3): 223–30. https://doi.org/10.17749/2077-8333/epi.par.con.2024.187.
7. Rinaldi B., Vaisfeld A., Amarri S., et al. Guideline recommendations for diagnosis and clinical management of Ring14 syndrome-first report of an ad hoc task force. Orphanet J Rare Dis. 2017; 12 (1): 69. https://doi.org/10.1186/s13023-017-0606-4.
8. Vaisfeld A., Spartano S., Gobbi G., et al. Chromosome 14 deletions, rings, and epilepsy genes: a riddle wrapped in a mystery inside an enigma. Epilepsia. 2021; 62 (1): 25–40. https://doi.org/10.1111/epi.16754.
9. Vasconcelos H.M. Jr., Vargas M.E., Pennesi M.E. Multimodal imaging of ring 14 syndrome associated maculopathy. Ophthalmic Genet. 2019; 40 (6): 541–4. https://doi.org/10.1080/13816810.2019.1688839.
10. Zollino M., Ponzi E., Gobbi G., Neri G. The ring 14 syndrome. Eur J Med Genet. 2012; 55 (5): 374–80. https://doi.org/10.1016/j.ejmg.2012.03.009.
11. Incecik F., Hergüner M.O., Mert G., et al. Ring chromosome 14 syndrome presenting with intractable epilepsy: a case report. Turk J Pediatr. 2013; 55 (5): 549–51.
12. Zampini L., Zanchi P., Rinaldi B., et al. Developmental trends of communicative skills in children with chromosome 14 aberrations. Eur J Pediatr. 2017; 176 (4): 455–64. https://doi.org/10.1007/s00431-017-2859-2.
13. Specchio N., Trivisano M., Serino D., et al. Epilepsy in ring 14 chromosome syndrome. Epilepsy Behav. 2012; 25 (4): 585–92. https://doi.org/10.1016/j.yebeh.2012.09.032.
14. Giovannini S., Marangio L., Fusco C., et al. Epilepsy in ring 14 syndrome: a clinical and EEG study of 22 patients. Epilepsia. 2013; 54 (12): 2204–13. https://doi.org/10.1111/epi.12393.
15. Unique. Understanding Rare Chromosome and Gene Disorders. Делеции длинного плеча 18-й хромосомы: от 18q21 до конца. URL: https://www.rarechromo.org/media/translations/Russian/18q%20deletions%20from%2018q21%20and%20beyond%20Russian%20FTNW.pdf (дата обращения 05.09.2024).
16. Jin Q., Qiang R., Cai B., et al. The genotype and phenotype of chromosome 18p deletion syndrome: case series. Medicine. 2021; 100 (18): e25777. https://doi.org/10.1097/MD.0000000000025777.
17. Pisano M., Sangiovanni G., D'Ambrosio F., et al. Oral care in a patient with long arm deletion syndrome of chromosome 18: a narrative review and case presentation. Am J Case Rep. 2022; 23: e936142. https://doi.org/10.12659/AJCR.936142.
18. Кузнецова М.А., Зрячкин Н.И., Елизарова Т.В. и др. Синдром del 18q у девочки-подростка 16 лет (обзор литературы с описанием клинического случая полиморфий и диагностических «коллизий»). Вестник Дагестанской государственной медицинской академии. 2021; 2: 39–47.
19. Hale D.E., Cody J.D., Baillargeon J., et al. The spectrum of growth abnormalities in children with 18q deletions. J Clin Endocrinol Metab. 2000; 85 (12): 4450–4. https://doi.org/10.1210/jcem.85.12.7016.
20. Versacci P., Digilio M.C., Sauer U., et al. Absent pulmonary valve with intact ventricular septum and patent ductus arteriosus: a specific cardiac phenotype associated with deletion 18q syndrome. J Med Genet A. 2005; 138A (2): 185–6. https://doi.org/10.102/ajmg.a.30916.
21. Cody J.D., Sebold C., Heard P., et al. Consequences of chromsome18q deletions. Am J Med Genet C Semin Med Genet. 2015; 169 (3): 265–80. https://doi.org/10.1002/ajmg.c.31446.
22. Zweier C., Sticht H., Bijlsma E.K. Further delineation of Pitt–Hopkins syndrome: phenotypic and genotypic description of 16 novel patients. J Med Genet. 2008; 45 (11): 738–44. https://doi.org/10.1136/jmg.2008.060129.
23. Котов А.С., Фирсов К.В., Санду Е.А. Фармакорезистентная эпилепсия. Клиническая лекция. Русский медицинский журнал. 2021; 6: 33–9.
24. Imataka G., Noguchi M., Tsukada K., et al. Partial epilepsy and developmental delay in infant with ring chromosome 14. Genet Couns. 2013; 24 (1): 81–3.
25. Новикова Л.Б., Акопян А.П., Шарапова К.М. и др. Эпилепсия у детей раннего возраста. Альманах молодой науки. 2022; 3: 26–28.
26. Новикова Л.Б., Акопян А.П., Шарапова К.М. и др. Особенности эпилепсии в раннем детском возрасте. Вестник Башкирского государственного медицинского университета. 2019; S1: 1548–54.
Новикова Лилия Бареевна, д.м.н., проф.
ул. Ленина, д. 3, г. Уфа 450008
Файзуллина Наиля Мухаметовна
ул. Ленина, д. 3, г. Уфа 450008
Акопян Анаит Погосовна, к.м.н., доцент
ул. Ленина, д. 3, г. Уфа 450008
Зюльцле Карина Маратовна, к.м.н., доцент
ул. Ленина, д. 3, г. Уфа 450008
Новикова Л.Б., Файзуллина Н.М., Акопян А.П., Зюльцле К.М. Неврологические проявления наследственных хромосомных заболеваний. Эпилепсия и пароксизмальные состояния. 2025;17(2):189-199. https://doi.org/10.17749/2077-8333/epi.par.con.2025.226
Novikova L.B., Faizullina N.M., Akopyan A.P., Ziultsle K.M. Neurological manifestations of hereditary chromosomal diseases. Epilepsy and paroxysmal conditions. 2025;17(2):189-199. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2025.226

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































