


Содержание
Перейти к:
 Н. А. Шнайдер,
Н. А. Пекарец,
Н. И. Пекарец,
Ю. Н. Быков,
В. В. Гречкина,
Д. В. Дмитренко,
М. М. Петрова,
Р. Ф. Насырова
Н. А. Шнайдер,
Н. А. Пекарец,
Н. И. Пекарец,
Ю. Н. Быков,
В. В. Гречкина,
Д. В. Дмитренко,
М. М. Петрова,
Р. Ф. Насырова https://doi.org/10.17749/2077-8333/epi.par.con.2025.239
Перейти к:
Актуальность. Метаболический синдром, индуцированный приемом противоэпилептических препаратов (ПЭП-МетС) является серьезной нежелательной реакцией (НР), которая снижает качество жизни больных эпилепсией и повышает риск коморбидных сердечно-сосудистых заболеваний, влияющих на продолжительность жизни. Риск возникновения данной НР варьируется в зависимости от различных факторов, которые определяют поиск чувствительных и специфичных биомаркеров для прогнозирования развития, профилактики, диагностики и коррекции ПЭП-МетС и его основных доменов (артериальная гипертензия, дислипидемия, центральное ожирение, сахарный диабет 2-го типа). Системный воспалительный ответ и оксидативный стресс – важные звенья как эпилептогенеза и нейродегенерации, так и патогенеза ПЭП-МетС.
Цель: систематизация результатов доклинических и клинических исследований роли микроРНК в развитии и неблагоприятном течении системного воспаления и оксидативного стресса, в эпилептогенезе и патогенезе ПЭП-МетС у пациентов с эпилепсией.
Материал и методы. Проведен анализ результатов фундаментальных и клинических исследований циркулирующих микроРНК как эпигенетических биомаркеров системных воспалительных реакций в механизме патогенеза МетС и ПЭП-МетС, поступивших в базы данных Google Scholar, PubMed/MEDLINE, MDPI, Scopus, и eLibrary за последнее десятилетие (2014–2024 гг.).
Результаты. Систематический обзор продемонстрировал, что микроРНК могут выступать как перспективные эпигенетические биомаркеры ПЭП-МетС, однако роль различных микроРНК и их паралогов в развитии данной НР вариабельна. В рамках настоящего исследования предложена сигнатура микроРНК в зависимости от риска развития и тяжести течения системного воспалительного ответа и ассоциированного с ним оксидативного стресса (ведущих механизмов патогенеза ПЭП-МетС). Данная сигнатура включает три группы микроРНК в зависимости от их роли в регуляции системного воспалительного ответа: низкий, средний, высокий риск.
Заключение. Роль микроРНК в регуляции системного воспалительного ответа при ПЭП-МетС нуждается в дальнейшем изучении и трансляции результатов фундаментальных исследований в реальную клиническую практику, поскольку рассмотренные микроРНК могут не только запускать и усугублять ПЭП-МетС, но и инициировать или поддерживать нейродегенеративные процессы, лежащие в основе эпилептогенеза.
Шнайдер Н.А., Пекарец Н.А., Пекарец Н.И., Быков Ю.Н., Гречкина В.В., Дмитренко Д.В., Петрова М.М., Насырова Р.Ф. Роль микроРНК как регуляторов системного воспалительного ответа при метаболическом синдроме, индуцированном противоэпилептическими препаратами. Эпилепсия и пароксизмальные состояния. 2025;17(2):208-226. https://doi.org/10.17749/2077-8333/epi.par.con.2025.239
Shnayder N.A., Pekarets N.A., Pekarets N.I., Bykov Yu.N., Grechkina V.V., Dmitrenko D.V., Petrova M.M., Nasyrova R.F. The role of microRNAs as regulators of systemic inflammatory response in anticonvulsant-induced metabolic syndrome. Epilepsy and paroxysmal conditions. 2025;17(2):208-226. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2025.239
По данным Всемирной организации здравоохранения, более 50 млн пациентов в мире страдают эпилепсией [1]. Ассоциированные с эпилепсией состояния приводят к снижению уровня жизни пациентов по сравнению со здоровыми людьми из-за риска травм во время приступов, нежелательных реакций (НР), возникающих при приеме противоэпилептических препаратов (ПЭП), и повышенной смертности, связанной с этим заболеванием [2].
Эпилептогенез является сложным многофакторным процессом, ассоциированным с изменениями в нейронных лигандах и потенциалзависимых ионных каналах, дисбалансом между возбуждающими и тормозящими нейромедиаторами, митохондриальной дисфункцией и активацией микроглии и макроглии (астроцитов) [3–5].
Системный воспалительный ответ, нарушающий гематоэнцефалический барьер (ГЭБ), играет одну из ключевых ролей в патогенезе нейродегенеративных заболеваний [6] и эпилептогенезе [4][5][7], дополнительно внося существенный вклад в развитие метаболического синдрома (МетС), индуцированного приемом ПЭП (ПЭП-МетС) [8]. Важным компонентом системного воспалительного ответа и нейровоспаления является окислительный стресс [9], который также отмечен в качестве одного из доменов ПЭП-МетС.
ПЭП представляют собой группу лекарственных средств (ЛС), которые используются для предотвращения и/или лечения эпилептических приступов путем контроля аномальной электрической активности в головном мозге. ПЭП применяются преимущественно для лечения эпилепсии, хотя могут назначаться пациентам с другими неврологическими заболеваниями и психическими расстройствами [10–12]. Больные эпилепсией принимают ПЭП длительно, иногда в течение всей жизни [13], что повышает риск развития НР со стороны различных органов и систем [14]. ПЭП-МетС является НР с негативными последствиями, включая повышение риска сердечно-сосудистых заболеваний, сахарного диабета 2-го типа и преждевременной смерти [15–17]. Однако это наименее изученная НР, поскольку в большинстве ранее проведенных исследований рассматривались отдельные домены ПЭП-МетС (повышение массы тела [18], абдоминальное ожирение [15], артериальная гипертензия [19], сахарный диабет 2-го типа [20] и др.), но не ПЭП-МетС в целом [15–18].
Известно, что риск ПЭП-МетС наиболее высок у пациентов, принимающих ПЭП первой генерации, включая карбамазепин (КБЗ) [15][21–26], фенитоин (ФТ) [15][20][22–27] и вальпроевую кислоту (ВК) [15][19–23][25–28]. Роль ПЭП новых генераций в развитии ПЭП-МетС пока недостаточно изучена, а их исследования одиночные и имеют противоречивые результаты, однако чаще демонстрируют протективные свойства (табл. 1) [15][17][25][26][29–32]. Это объясняет повышение интереса исследователей и клиницистов к проблеме ПЭП-МетС и поиску новых биомаркеров данной НР, включая клинические, метаболические, генетические и эпигенетические [19][33][34].
Таблица 1. Роль противоэпилептических препаратов в развитии различных компонентов метаболического синдрома
Table 1. The role of antiepileptic drugs in the development of various components of the metabolic syndrome
|
ПЭП / AED |
Эффект / Effect |
Источники / References |
|
Первые генерации / First generations |
||
|
Фенобарбитал / Phenobarbital |
Дислипидемия / Dyslipidemia Cистемное воспаление и оксидативный стресс / Systemic inflammation and oxidative stress Центральное ожирение и/или повышение массы тела // Central obesity and/or weight gain |
[22][25][27][32] |
|
Вальпроевая кислота и ее соединения / Valproic acid and compounds |
Cистемное воспаление и оксидативный стресс / Systemic inflammation and oxidative stress Дислипидемия / Dyslipidemia Центральное ожирение и/или повышение массы тела // Central obesity and/or weight gain Вариабельный эффект на артериальное давление / Variable effect on blood pressure Предположительно препятствует развитию инсулинорезистентности и СД2 / Presumably prevents the development of insulin resistance and T2DM |
[15][19–23][25–28] |
|
Фенитоин / Phenytoin |
Дислипидемия / Dyslipidemia Cистемное воспаление и оксидативный стресс / Systemic inflammation and oxidative stress Гипергликемия / Hyperglycemia Центральное ожирение и/или повышение массы тела // Central obesity and/or weight gain |
[15][20][22–27] |
|
Карбамазепин / Carbamazepine |
Дислипидемия / Dyslipidemia Cистемное воспаление и оксидативный стресс / Systemic inflammation and oxidative stress Центральное ожирение и/или повышение массы тела // Central obesity and/or weight gain |
[15][21–26] |
|
Бензодиазепины / Benzodiazepines |
Повышение массы тела / Weight gain Снижение секреции инсулина / Decreased insulin secretion Ингибирование системного воспалительного ответа / Inhibition of systemic inflammation |
[25][26][35][36] |
|
Этосуксимид / Ethosuximide |
Повышение массы тела / Weight gain |
[25] |
|
Новые генерации / Next generations |
||
|
Ламотриджин / Lamotrigine |
Антиоксидативный эффект / Antioxidant effect Снижение массы тела / Weight loss |
[15][25][30][31] |
|
Леветирацетам, бриварацетам / Levetiracetam, brivaracetam |
Преимущественно антиоксидативный эффект, однако имеются сведения об окислении ДНК, липидов и глутатиона / Predominantly antioxidant effect, evidence of DNA, lipids and glutathione oxidation Ингибирование системного воспалительного ответа / Inhibition of systemic inflammation |
[26][30][31][37] |
|
Габапентин, прегабалин / Gabapentin, рregabalin |
Нейропротективный эффект / Neuroprotective effect Антиоксидативный эффект / Antioxidant effect Повышение массы тела / Weight gain Потенциальное снижение уровня глюкозы в крови / Potential reduction of blood glucose levels |
[15][25][30][32] |
|
Окскарбазепин / Oxcarbazepine |
Антиоксидативный эффект / Antioxidant effect Повышение массы тела / Weight gain |
[25][31] |
|
Тиагабин / Tiagabin |
Антиоксидативный эффект в низких дозах / Antioxidant effect at low doses Прооксидативный эффект в высоких дозах / Pro-oxidant effect at high doses |
[30] |
|
Вигабатрин / Vigabatrin |
Системное воспаление и оксидативный стресс / Systemic inflammation and oxidative stress Центральное ожирение и/или повышение массы тела / Central obesity and/or weight gain |
[15][26][30] |
|
Фелбамат / Felbamat |
Антиоксидативный эффект / Antioxidant effect |
[30] |
|
Топирамат / Topiramate |
Антиоксидативный эффект / Antioxidant effect Снижение массы тела / Weight loss Препятствует развитию гипергликемии и инсулинорезистентности / Prevents the hyperglycemia and insulin resistance |
[15][25][30][32] |
|
Лакосамид / Lacosamide |
Повышение массы тела / Weight gain |
[25] |
|
Зонисамид / Zonisamide |
Снижение массы тела / Weight loss |
[15][25] |
|
Перампанел / Perampanel |
Повышение массы тела / Weight gain |
[25] |
Примечание: ПЭП – противоэпилептический препарат; СД2 – сахарный диабет 2-го типа; ДНК – дезоксирибонуклеиновая кислота.
Note. AED – antiepileptic drug; T2DM – type 2 diabetes mellitus; DNA – deoxyribonucleic acid.
Биомаркер представляет собой определенную характеристику, которая отражает как нормальные биологические, так и патологические процессы, а также результаты реакций на различные воздействия и интервенции и подходит для целей диагностики, мониторинга, объективной оценки риска развития патологических состояний и прогнозирования их исходов. Биомаркеры используются и Для оценки безопасности ЛС, применяемых в эпилептологии, и риска возникновения НР [38][39], включая ПЭП-МетС [15–27][29–32].
На основе исследования лабораторных биомаркеров предложено диагностировать три варианта ПЭП-МетС у детей и взрослых с эпилепсией и эпилептическими синдромами, получающих ПЭП в течение 3 мес и более, в соответствии с действующими критериями Международной диабетической федерации (англ. International Diabetes Federation, IDF) и Отчета по лечению взрослых III (англ. Adult Treatment Panel III, ATP III) Национальной образовательной программы по холестерину (англ. National Cholesterol Education Program): определенный, возможный и вероятный [40]. Следует отметить, что отсутствие клинических и лабораторных биомаркеров МетС у пациентов с эпилепсией и/или эпилептическими синдромами в течение 3 мес от старта терапии ПЭП не исключает вероятность развития ПЭП-МетС в будущем, если прием препаратов продолжается в дальнейшем. Важен динамический контроль этих биомаркеров у больных с возможным ПЭП-МетС – 1 раз в 3 мес, с вероятным ПЭП-МетС – 1 раз в 6 мес [19].
Чувствительность и специфичность метаболических (биохимических и гормональных) биомаркеров ПЭП-МетС могут варьироваться в широком диапазоне в зависимости от влияния внешнесредовых факторов (например, климатогеографических, культуральных, пищевых), а также от возраста и пола пациентов с эпилепсией [19]. Кроме того, на чувствительность и специфичность метаболических биомаркеров ПЭП-МетС могут оказывать влияние условия забора и хранения образцов. Это побуждает исследователей к поиску новых биомаркеров, которые обладали бы лучшим профилем стабильности в образцах, а также хорошей воспроизводимостью результатов исследования в различных лабораториях.
В последние годы активно изучаются биомаркеры, связанные с геномом и эпигеномом человека [41–44], в особенности эпигенетические, не зависящие от изменения в последовательности ДНК, а регулирующие экспрессию тех или иных генов в зависимости от воздействия факторов внешней среды, включая климатогеографические условия проживания, особенности питания. Также принимаемые ЛС могут влиять на то, какая именно часть генома человека будет отключена или включена в определенные периоды жизни в норме и патологии. Способные воспроизводиться в течение всего цикла деления клеток эпигенетические биомаркеры не зависят от изменения в последовательности ДНК, хотя влияют на клеточные и физиологические фенотипические признаки организма человека или даже могут быть причиной развития эпилепсии и ПЭП-МетС [45][46].
Перспективными эпигенетическими биомаркерами ПЭП-МетС являются короткие некодирующие одноцепочечные РНК длиной 19–25 нуклеотидов – микроРНК [47–52]. МикроРНК способны регулировать структуру хроматина, воздействуя на ключевые модификаторы гистонов и метилирование ДНК, снижая или повышая риск развития ПЭП-МетС [53].
Цель – систематизация результатов доклинических и клинических исследований роли микроРНК в развитии и неблагоприятном течении системного воспаления и оксидативного стресса, в эпилептогенезе и патогенезе ПЭП-МетС у пациентов с эпилепсией.
Проведен анализ результатов доклинических и клинических исследований микроРНК как эпигенетических биомаркеров системного воспалительного ответа в механизме патогенеза МетС и ПЭП-МетС, поступивших в базы данных Google Scholar, PubMed/MEDLINE, MDPI, Scopus и eLibrary за последнее десятилетие (2014–2024 гг.). Обзор выполнен в соответствии с рекомендациями PRISMA (англ. Preferred Reporting Items for Systematic reviews and Meta-Analyses) [54].
Поиск осуществляли по ключевым словам и их комбинациям на русском («эпилепсия», «метаболический синдром», «ожирение», «дислипидемия», «артериальная гипертензия», «гипергликемия», «микроРНК», «противоэпилептические препараты», «системное воспаление», «оксидативный стресс») и английском (“epilepsy”, ”arterial hypertension”, “hyperglycemia”, “microRNA”, “antiepileptic drugs”, “systemic inflammation”, “oxidative stress”) языках.
Критериями включения публикаций в обзор являлись:
– тип доступа (открытый доступ к полнотекстовой версии);
– язык (публикации на русском или английском языке);
– тип публикации (оригинальная статья, систематический обзор, метаанализ, кокрейновский обзор).
Критерии исключения:
– дублирующие публикации;
– постеры;
– материалы конференций;
– диссертации и авторефераты диссертаций;
– материалы, опубликованные на правах рукописи.
Изменение уровня экспрессии анализируемых микроРНК оценивалось в биологических жидкостях (крови, плазме, сыворотке, экзосомах, мононуклеарах) пациентов и на животных моделях в качестве биомаркеров развития МетС или ПЭП-МетС, а также их основных доменов (системного воспалительного ответа и оксидативного стресса).
Системное воспаление является чрезмерной защитной реакцией организма, которая характеризуется повышением сывороточных уровней белков острой фазы и провоспалительных цитокинов, включая С-реактивный белок (СРБ), фактор некроза опухоли альфа (ФНО-α), интерлейкин 1-бета (ИЛ-1β), ИЛ-6, ИЛ-8 и ИЛ-17, а также инфильтрацией тканей макрофагами и Т-лимфоцитами [55][56].
Системное воспаление и эпилептогенез
В ходе многочисленных исследований как in vivo, так и in vitro была подтверждена важная роль воспалительных медиаторов и активированных иммунокомпетентных клеток в эпилептогенезе и разрушении ГЭБ [4][7]. Эпилептогенные события (инсульты, инфекции центральной нервной системы (ЦНС), гипоксия и другие повреждения тканей головного мозга), равно как и сами эпилептические приступы могут спровоцировать воспалительные процессы, повышая экспрессию провоспалительных цитокинов и их родственных рецепторов, например белка высокой подвижности 1-й группы (англ. high-mobility group protein B1, HMGB1) и ИЛ-1β, которые высвобождаются из активированных глиальных клеток и стимулированных нейронов после эпилептических приступов, а затем связываются с толл-подобными рецепторами (англ. toll-like receptor, TLR) и рецептором ИЛ-1. В результате нейровоспаления активированные астроциты и микроглия начинают вырабатывать и высвобождать другие провоспалительные молекулы, что усиливает последующие воспалительные каскадные реакции [4][5].
После воздействия патогенных факторов нейроны продолжают выделять медиаторы воспаления, такие как белки теплового шока (англ. heat shock proteins, HSP), гиалуронан, нуклеиновые кислоты, гепарансульфат, сурфактант-A, фибриноген, аденозинтрифосфат (АТФ) и HMGB1. Далее цитокины распознаются микроглией через рецепторы распознавания паттернов (РРП), что способствует поддержанию воспалительных реакций в ЦНС даже при отсутствии инфекции или других повреждающих факторов (например, гипоксии) [57].
Длительное воспаление может привести к повреждению ГЭБ, через который провоспалительные цитокины и иммунные клетки из периферической крови проникают в головной мозг и участвуют в эпилептогенезе. Избыточная продукция медиаторов воспаления вызывает аномальную гиперактивность нейронов, что приводит к увеличению проницаемости глутаматергических нейронов для ионов кальция (Ca²⁺), вызывая явления эксайтотоксичности [7].
HMGB1 играет провоспалительную роль, активируя основной РРП: TLR4, который, в свою очередь, запускает провоспалительный каскад, активируя медиатор миелоидной дифференцировки 88-го типа (англ. myeloid differentiation primary response gene, MyD88), митоген-активированную протеинкиназу (англ. mitogen-activated protein kinase, МАРК) и ядерный фактор каппа В (англ. nuclear factor kappa B, NF-κB) [4][58][59]. Активированный TLR4 усиливает приток ионов Ca²⁺ в нейроны посредством рецептора N-метил-D-аспартата (англ. N-methyl-D-aspartate, NMDA), обусловливая явления эксайтотоксичности и индуцируя эпилептогенез [60].
АТФ является главной молекулярной единицей накопления энергии, а также одним из возбуждающих медиаторов ЦНС. Внеклеточное ее содержание обычно сохраняется на достаточно низком уровне, однако активная выработка АТФ нейрональными клетками и глией во внеклеточное пространство, например после эпилептических приступов, в состоянии гипоксии или после иного клеточного повреждения, ассоциировано с высвобождением HMGB1 посредством активации NOD-подобного рецепторного белка 3 (англ. NOD-like-receptor protein 3, NLRP3) инфламмасомы и пуринергического рецептора P2X7R, что усугубляет течение воспалительной реакции в ЦНС, повышая экспрессию провоспалительных ИЛ-1β и ИЛ-6 [61][62]. Ось АТФ/P2X7R тоже играет роль в увеличении количества активных форм кислорода (АФК) в микроглии и высвобождении простагландина E2 (ПГЕ2) [5][58], а также индуцирует развитие глиоза и астроглиоза, разрушающих ГЭБ [4]. В подтверждение этой гипотезы подавление окислительного стресса низкомолекулярным каталитическим антиоксидантом MnIIITDE-2-ImP⁵⁺ приводило к снижению выработки провоспалительных цитокинов и активации микроглии [58].
Как известно, после первого эпилептического приступа повышается экспрессия провоспалительных цитокинов, в частности ИЛ, а также их рецепторов [63]. Так, установлено, что ИЛ-1β, выделяемый активированной астроглией и нейронами после первого приступа, связывается с рецептором 1-го типа к ИЛ-1 (ИЛ-1R1) и усиливает экспрессию медиаторов ИЛ-6, ФНО-α и циклооксигеназы 2-го типа (ЦОГ-2) посредством микроРНК (miR-146a) [5][64]. Система ИЛ-1β/ИЛ-1R1 фосфорилирует NMDA-рецепторы, усиливая приток в них ионов Ca²⁺, а также уменьшает высвобождение гамма-аминомасляной кислоты (ГАМК) в синапсах, активируя MyD88 и способствуя развитию эпилепсии вследствие нарушения равновесия в системе ГАМК/глутамат [4].
ИЛ-6 и его рецептор (ИЛ-6R) также экспрессируются астроцитами и нейронами после эпилептических приступов, однако они могут оказывать как эпилептогенные, так и противоэпилептические эффекты в зависимости от своей концентрации и продолжительности экспозиции в ЦНС и ассоциированы с изменением плотности постсинаптических ГАМК-рецепторов и повышением чувствительности рецепторов NMDA и α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислоты (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid, AMPA) к глутамату [65][66].
Зависящим от концентрации эффектом на эпилептогенез обладает также ФНО-α, оказывая эпилептогенное действие на нейроны гиппокампа в высоких дозах, связываясь с рецептором p55ФНО, а противоэпилептическое действие он имеет при низкой концентрации посредством связи с рецептором p75ФНО [4]. Эпилептогенный эффект ФНО-α может быть ассоциирован с увеличением проницаемости AMPA- и NMDA-рецепторов для ионов Ca²⁺ после связи с p55ФНО, этот же механизм объясняет ФНО-α-индуцированную эксайтотоксичность [67], играющую известную роль в эпилептогенезе.
Трансформирующий фактор роста бета (TФР-β) вызывает быструю регуляцию и секрецию ИЛ-6, усиливая специфическое для астроцитов фосфорилирование smad-белков и тем самым вызывая нейронную эксайтотоксичность и эпилептические приступы. Кроме того, TФР-β стимулирует поглощение альбумина астроцитами. Это приводит к снижению экспрессии белков щелевых контактов – коннексинов. В результате нарушается буферизация ионов K⁺ и глутамата в астроцитах. Как следствие, ионы K⁺ накапливаются в синаптической щели. Это может активировать NMDA-рецепторы и вызвать деполяризацию постсинаптической мембраны [68].
Фактор активации тромбоцитов (ФАТ) играет важную роль в развитии воспалительных реакций. Он способствует увеличению концентрации ионов Ca²⁺ в нейронах и астроцитах, стимулирует высвобождение глутамата и подавляет активность ГАМК-рецепторов. Кроме того, ФАТ участвует в модуляции синаптической передачи сигналов, активируя сигнальные пути транскрипции ЦОГ-2 [69].
Сама же ЦОГ-2, обычно экспрессируемая в ЦНС на низком уровне, но способная повышаться вследствие черепно-мозговых травм, инсультов, а также повторных эпилептических приступов, является важным медиатором воспаления и вызывает отсроченное повреждение ГАМКергических нейронов после судорог в моделях височной эпилепсии у грызунов. Однако важно упомянуть, что использование ингибиторов ЦОГ-2 (рефококсиб, целекоксиб) с целью снижения нейровоспаления и порога судорожной готовности дало противоречивые результаты: на животных моделях до судорог оно существенно увеличивало смертность грызунов после судорог, вызванных каиновой кислотой [7][70], а их применение после эпилептического приступа существенно снижало смертность, а также уменьшало возбудимость постсинаптической мембраны и обратный приток ионов Ca²⁺, связанный с потенциалом действия дендритов в гранулярных нейронах на моделях височной эпилепсии у мышей. Однако это же исследование подтвердило неспособность селективных ингибиторов ЦОГ-2 предотвратить развитие эпилептогенеза или снизить частоту рецидивов судорог на животных моделях эпилепсии [71].
Продуктом ЦОГ-2 является ПГЕ2, который, в свою очередь, может увеличивать высвобождение глутамата астроцитами и вызывать перевозбуждение нейронов [7].
Системное воспаление и ПЭП-МетС
Непосредственное влияние ПЭП на развитие МетС и его компонентов подтверждено в многочисленных исследованиях [15–27][29–32]. Так, ФТ связан с повышенным уровнем глюкозы и снижением секреции инсулина, однако ВК, несмотря на частое увеличение массы тела [28], наоборот стимулировала секрецию инсулина β-клетками островков Лангерганса поджелудочной железы [20]. Такие домены ПЭП-МетС, как центральное ожирение, дислипидемия и гипергликемия, ассоциированы с системным воспалительным ответом, усугубляя тем самым течение эпилепсии и обусловливая развитие терапевтически резистентной эпилепсии [17][22][25][27][30].
ПЭП влияют на синтез провоспалительных цитокинов сложным образом. ВК показала противоречивые результаты, снижая in vitro выработку ФНО-α и ИЛ-6, предположительно за счет ингибирования NF-κB. При этом в ходе клинических исследований ВК повышала уровни ИЛ-1, ИЛ-6 и ИЛ-5 у пациентов с эпилепсией. КБЗ стимулирует выработку провоспалительных цитокинов ИЛ-1α, ИЛ-1β, ИЛ-2, ИЛ-10 и TФР-β in vitro и в ряде случаев повышает уровни иммуноглобулинов (англ. immunoglobulin, Ig) и натуральных киллеров (англ. natural killer (NK) cells). ФТ повышает уровень ИЛ-1 и высвобождение TФР-β, снижает уровни IgA и Т-супрессоров у пациентов с эпилепсией. КБЗ и ФТ также повышают уровни СРБ, в то время как ламотриджин (ЛТД) и ВК снижают его концентрацию в плазме крови [72]. Клоназепам, как и другие бензодиазепины, демонстрировал преимущественно противовоспалительные свойства, подавляя выработку ИЛ-6 и препятствуя миграции активированных макрофагов и нейтрофилов в ЦНС [35]. Диазепам также уменьшает системное воспаление, подавляя активность Т-лимфоцитов, нейтрофилов и NK-клеток, а также выработку гамма-интерферона [26].
Леветирацетам (ЛЕВ) оказывает преимущественно противовоспалительное действие путем ингибирования ИЛ-1β в гиппокампе [26][73].
Результаты исследований взаимосвязей между приемом ПЭП и изменением уровней биомаркеров системного воспалительного ответа в крови противоречивы и демонстрируют как провоспалительные (ПЭП ранних генераций), так и противовоспалительные (ПЭП новых генераций) эффекты, поэтому их роль в развитии ПЭП-МетС дискутабельна и во многом зависит от поколения ПЭП [73–75].
Маркеры системного воспалительного ответа
Как указано выше, системный воспалительный ответ является важным звеном эпилептогенеза и МетС и может индуцироваться, увеличивать интенсивность и поддерживаться при приеме ПЭП, что обусловливает важность ранней идентификации его биомаркеров для оценки рисков развития НР и коррекции терапии (табл. 2).
Таблица 2. Биомаркеры системного воспаления
Table 2. Biomarkers of systemic inflammation
|
Биомаркер / Biomarker |
Роль в патогенезе / Role in pathogenesis |
Источники / References |
|
HMGB1 |
Активация микроглии и астроцитов / Activation of microglia and astrocytes Повышение экспрессии факторов транскрипции медиаторов воспаления / Increased expression of inflammatory mediator transcription factors |
[4] |
|
АТФ / ATP |
Высвобождение HMGB1 из инфламосом / HMGB1 release from inflammosomes Высвобождение провоспалительных ИЛ-1β и ИЛ-6 / Pro-inflammatory IL-1β and IL-6 release Повышение концентрации АФК в микроглии и высвобождение ПГЕ2 / Increased ROS concentration in microglia and PGE2 release Индукция глиоза в ЦНС / Gliosis induction in CNS |
[4][5][58][61][62] |
|
Система ИЛ-1β/ИЛ-1R1 // IL-1β/IL-1R1 system |
Повышение экспрессии провоспалительных ИЛ-6, ФНО-α и ЦОГ-2 / Increased expression of proinflammatory IL-6, TNF-α and COX-2 Фосфорилирование NMDA-рецепторов и повышение притока ионов Ca²⁺ в глутаматергические нейроны / Phosphorylation of NMDA receptors and increased influx of Ca²⁺ ions into glutamatergic neurons Уменьшение высвобождения ГАМК в ГАМКергических нейронах / Reduction of GABA release in GABAergic neurons |
[4][5][62][64] |
|
Система ИЛ-6/ИЛ-6R // IL-6/IL-6R system |
Снижение плотности постсинаптических рецепторов ГАМК / Decreased density of postsynaptic GABA receptors Увеличение чувствительности глутаматергических рецепторов AMPA и NMDA / Increased sensitivity of AMPA and NMDA glutamat receptors |
[65][66] |
|
СРБ / CRP |
Активация микроглии и астроцитов / Activation of microglia and astrocytes Повышение экспрессии факторов транскрипции медиаторов воспаления / Increased expression of inflammatory mediator transcription factors |
[72] |
|
ФНО-α / TNF-α |
Повышение проницаемости AMPA- и NMDA-рецепторов глутамата для ионов Ca²⁺ / Increased permeability of AMPA and NMDA glutamate receptors for Ca²⁺ ions |
[67] |
|
TФР-β / TGF-β |
Повышение экспрессии ИЛ-6 / Increased IL-6 expression Повышение поглощения альбумина астроцитами с нарушением функции коннексинов и ростом концентрации ионов К⁺ в синаптической щели глутаматергических нейронов / Increased albumin uptake by astrocytes with impaired connexin function and increased concentration of K⁺ ions in the synaptic cleft of glutamatergic neurons |
[68] |
|
ФАТ / PAF |
Повышение концентрации ионов Ca²⁺ в нейронах и астроцитах / Increased concentration of Ca²⁺ ions in neurons and astrocytes Снижение активности ГАМК-рецепторов / Decreased GABA receptor activity Повышение высвобождения глутамата в синаптическую щель / Increased glutamate release into the synaptic cleft Повышение экспрессии ЦОГ-2 / Increased COX-2 expression |
[69] |
|
ЦОГ-2 / COX-2 |
Повышение притока ионов Ca²⁺ в глутаматергические нейроны с их последующим возбуждением / Increased influx of Ca²⁺ ions into glutamatergic neurons with their subsequent excitation Повреждение ГАМК-нейронов / GABA neurons damage Синтез ПГЕ2 / PGE2 synthesis |
[7][70][71] |
|
ПГЕ2 / PGЕ2 |
Увеличение высвобождения глутамата астроцитами / Increased glutamate release by astrocytes Перевозбуждение нейронов вследствие повышения тока ионов Ca²⁺ / Neuron overexcitation due to increased Ca²⁺ ion current |
[7] |
Примечание. HMGB1 (англ. high-mobility group protein B1) – белок высокой подвижности 1-й группы; АТФ – аденозинтрифосфат; ИЛ – интерлейкин; СРБ – С-реактивный белок; ФНО-α – фактор некроза опухоли альфа; ТФР-β – трансформирующий фактор роста бета; ФАТ – фактор активации тромбоцитов; ЦОГ-2 – циклооксигеназа-2; ПГЕ2 – простагландин E2; АФК – активные формы кислорода; ЦНС – центральная нервная система; NMDA (англ. N-methyl-D-aspartate) – N-метил-D-аспартат; ГАМК – гамма-аминомасляная кислота; AMPA (англ. α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) – α-амино-3-гидрокси-5-метил-4-изоксазолпропионовой кислота.
Note. HMGB1 – high-mobility group protein B1; ATP – adenosine triphosphate; IL – interleukin; CRP – C-reactive protein; TNF-α – tumor necrosis factor alpha; TGF-β – transforming growth factor beta; PAF – platelet activating factor; COX-2 – cyclooxygenase-2; PGE2 – prostaglandin E2; ROS – reactive oxygen species; CNS – central nervous system; NMDA – N-methyl-D-aspartate; GABA – gamma-aminobutyric acid; AMPA – α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid.
Оксидативный стресс представляет собой дисбаланс между повышенным уровнем АФК и низкой активностью антиоксидантных механизмов. АФК являются важными компонентами митохондриального β-окисления, но их избыток может приводить к непосредственному повреждению нейронов. Высокие уровни АФК физиологичны при чрезмерных физических нагрузках, а также являются следствием процессов, протекающих при естественном старении организма и нейродегенерации. В условиях эпилептогенеза повышение уровней АФК и, как следствие, развитие оксидативного стресса, оказывает негативный эффект, поддерживающий системное воспаление и активирующий механизмы нейродегенерации [76].
Оксидативный стресс и эпилептогенез
АФК играют ключевую роль в эпилептогенезе, провоцируя повреждение нейронов, вызванное эпилептическими приступами, и окисляя ионные каналы и переносчики тормозных нейромедиаторов. Это, в свою очередь, приводит к повышению возбудимости нейронов, изменению потенциала действия плазматической мембраны и снижению активности тормозных интернейронов в головном мозге [77].
Эпилепсия сама по себе способствует оксидативному стрессу. Многочисленные экспериментальные и клинические исследования подтверждают повышенное потребление кислорода и глюкозы головным мозгом уже после первого эпилептического приступа [5][22][78].
Также после первого эпилептичсекого приступа микроглия начинает участвовать в процессах нейровоспаления и нейродегенерации, активируя комплекс никотинамидадениндинуклеотидфосфат (НАДФ) оксидазы (англ. nicotinamide adenine dinucleotide phosphate oxidase, NOX), и увеличивает выработку АФК [79]. Повышение уровня АФК происходит по той же схеме, что и высвобождение провоспалительных цитокинов: инактивация митохондриального комплекса I типа, посттрансляционная модификация и нарушение митохондриального дыхания в животных моделях височной эпилепсии [58]. Высокие уровни АФК в ЦНС активируют сигнальный путь NF-κB и вызывают высвобождение HMGB1, влияние которых на эпилептогенез освещалось выше [4].
ЦНС особенно чувствительна к повреждающему воздействию АФК, поскольку активность антиоксидативных ферментов в головном мозге примерно в 10 раз ниже, чем в других тканях организма человека. Следовательно, образующиеся свободные АФК могут окислять и нарушать функцию тормозных нейромедиаторов и нейронных мембран, что приводит к снижению их текучести и нарушению мембранного транспорта [5].
ЦНС потребляет до 20% вдыхаемого кислорода, при этом бо́льшая его часть используется в нейрональных митохондриях для синтеза АТФ в процессе клеточного дыхания, при котором выделяется значительное количество АФК и оксида азота (NO) [80]. Митохондриальная дисфункция, вызванная окислительным стрессом, играет заметную роль в нейродегенерации и эпилептогенезе. Так, повторяющиеся эпилептические приcтупы провоцируют чрезмерную выработку АФК в митохондриях, которые в присутствии ионов Cu²⁺ и Fe²⁺ окисляют липиды, белки и ДНК. Это приводит к нарушению энергетического метаболизма в нейронах и изменению экспрессии генов, ассоциированных с повышением проницаемости мембран и возбудимости нейронов, что в конечном итоге снижает порог судорожной готовности [78]. Митохондрии также поддерживают стабильный внутриклеточный уровень ионов Cа²⁺, поэтому митохондриальная дисфункция приводит к его избытку. Совместное воздействие АФК и избытка ионов Cа²⁺ усиливает синаптическую передачу, возбудимость нейронов и эксайтотоксичность. В результате открываются поры перехода проницаемости митохондрий и происходит перемещение проапоптотических молекул в цитозоль нейронов, что инициирует их апоптоз [5].
В случае посттравматической эпилепсии высвобождение ионов Fe³⁺ из эритроцитов приводит к выработке супероксида, гидроксида и пероксида водорода. Перекисное окисление липидов (ПОЛ), вызванное избыточной продукцией АФК, и последующая деградация мембран нейронов сопровождаются повышенной секрецией возбуждающих аминокислот. Кроме того, гидроксильные радикалы способствуют выработке метилгуанидина – эндогенного проконвульсанта, образующегося из креатинина [81].
Избыточное образование АФК повреждает митохондриальную ДНК и снижает активность ферментов цикла Кребса. Вызванные АФК изменения в структуре глутаматных рецепторов, снижение энергозависимого действия переносчиков глутамата и потеря ГАМКергических нейронов повышают возбудимость нейронов и их предрасположенность к судорогам [78].
Исходя из важности окислительного стресса в эпилептогенезе, активно обсуждается потенциальная роль антиоксидантов в качестве болезнь-модифицирующей терапии эпилепсии и ПЭП-МетС, в особенности препаратов мелатонина, α-токоферола и коэнзима Q10 [5][80]. Однако применение экзогенного мелатонина и агонистов мелатониновых рецепторов при эпилепсии нуждается в дальнейшем изучении. Эндогенный мелатонин может действовать как проконвульсант из-за снижения дофаминергической передачи в головном мозге, в то время как экзогенный мелатонин или его агонисты (агомелатин) индуцировали ГАМКергическое торможение в ЦНС [80].
Оксидативный стресс и ПЭП-МетС
Окислительный стресс и системное воспаление играют важную роль в развитии метаболических коморбидных нарушений, сопутствующих заболеваний (гиперлипидемия, артериальная гипертензия и сниженная толерантность к глюкозе) у пациентов с эпилепсией [82–84].
АФК участвуют в развитии дезадаптивных реакций, которые приводят к МетС и системному воспалительному ответу разной степени выраженности в зависимости от типа клеток и тканевой среды [85][86]. Избыточная выработка АФК, наблюдаемая при ожирении, инсулинорезистентности, гипергликемии и дислипидемии, доказывает взаимосвязь оксидативного стресса и ПЭП-МетС [83][87][88]. В многочисленных исследованиях установлено, что у людей с ПЭП-МетС снижается активность антиоксидантных ферментов в крови и повышаются уровни биомаркеров окислительного повреждения [37][82][83][85][87].
Имеются многочисленные клинические и экспериментальные отчеты, демонстрирующие ПЭП-индуцированные изменения оксидантно-антиоксидантного баланса у пациентов с эпилепсией. ПЭП первой генерации (фенобарбитал (ФБ), ФТ, ВК) активируют ПОЛ и образование АФК у пациентов с эпилепсией при длительной фармакотерапии, что индуцирует повышение уровней прооксидативных ферментов (супероксиддисмутазы и каталазы), одновременно понижая уровни антиоксидативных ферментов (глутатионпероксидазы и глутатионредуктазы) [62]. Так, ВК достоверно снижает уровни антиоксидативного фермента сиртуина 1-го типа (англ. sirtuin 1, SIRT1) в крови [66]. Известно, что реактивные метаболиты некоторых ПЭП первой генерации (ФБ, ФТ, КБЗ, ВК) могут вызывать образование АФК и способствуют повреждению клеток [22][89]. В целом ПЭП первой генерации не имеют существенного нейропротективного и антиоксидативного эффекта.
По сравнению с ними, ПЭП новых генераций, например ЛТД, снижали ПОЛ и уровни АФК, в то время как окскарбазепин существенно уменьшал уровни биомаркеров окислительного стресса у взрослых пациентов с эпилепсией [63][64]. В отношении ЛЕВ имеются противоречивые результаты, согласно которым у больных, принимающих данный ПЭП, могут повышаться уровни окисленных липидов (малоновый диальдегид) и ДНК (8-оксо-2'-дезоксигуанозин), а также снижаться уровни восстановленного глутатиона [90][91]. Некоторая антиоксидативная активность наблюдается также у габапентина и зонисамида, способных подавлять ПОЛ, снижая выработку NO и поглощая гидроксильные радикалы, а также повышая активность ферментов антиоксидативной системы (каталаза и глутатион) [5][80].
Исследования вигабатрина и тиагабина показали противоречивые результаты. Вероятно, эти ПЭП имеют дозозависимый прооксидантный эффект, поскольку в ряде работ они повышали уровни АФК при достижении субтоксических и токсических концентраций в крови по данным терапевтического лекарственного мониторинга [30].
Маркеры оксидативного стресса
Выполненный нами анализ ранее проведенных доклинических и клинических исследований позволил систематизировать перспективные биомаркеры оксидативного стресса как ведущего домена ПЭП-МетС, играющего важную роль в инициации и поддержании системного воспалительного ответа и нейровоспаления (табл. 3).
Таблица 3. Биомаркеры оксидативного стресса как ведущего домена метаболического синдрома, индуцированного противоэпилептическими препаратами
Table 3. Biomarkers of oxidative stress as a leading domain of antiepileptic drug-induced metabolic syndrome
|
Биомаркер / Biomarker |
Роль в патогенезе / Role in pathogenesis |
Источники / References |
|
АФК (HO, HClO, O2–, H2O2), NO / ROS (HO, HClO, O2–, H2O2), NO |
Высвобождение HMGB1 из инфламосом / HMGB1 release from inflammosomes Окисление нейромедиаторов и мембран нейронов с нарушением трансмембранного транспорта / Oxidation of neurotransmitters and neuron membranes with impaired transmembrane transport Окисление нейрональных липидов, белков и ДНК / Oxidation of neuronal lipids, proteins, and DNA Развитие митохондриальной дисфункции c активацией проапоптических молекул / Development of mitochondrial dysfunction with activation of pro-apoptotic molecules Повышение возбудимости и синаптической передачи поврежденных нейронов / Increased excitability and synaptic transmission of damaged neurons Повышение уровней факторов транскрипции прооксидативных ферментов (NF-κB) / Increased levels of pro-oxidative enzyme transcription factors (NF-κB) |
[4][5][78] |
|
Прооксидативные ферменты (НАДФ(Н)оксидаза, миелопероксидаза, ксантиноксидаза, синтаза оксида азота) / Pro-oxidative enzymes (NADP(H) oxidase, myeloperoxidase, xanthine oxidase, nitric oxide synthase) |
Повышение уровней АФК и NO / Increased ROS and NO levels |
[79] |
|
Дисульфид глутатиона / Glutathione disulfide |
Биомаркер истощения антиоксидативной системы глутатиона / Biomarker of glutathione antioxidant system depletion |
[86][92] |
|
Антиоксидативные ферменты (каталаза, супероксиддисмутаза, глутатионпероксидаза, глутатионредуктаза) / Antioxidant enzymes (catalase, superoxide dismutase, glutathione peroxidase, glutathione reductase) |
Биомаркер общего истощения антиоксидативной системы организма / Biomarker of general depletion of the body's antioxidant system |
[93] |
|
Неферментные антиоксиданты (мочевая кислота, билирубин, токоферол, аскорбиновая кислота, каратиноиды) / Non-enzymatic antioxidants (uric acid, bilirubin, tocopherol, ascorbic acid, carotenoids) |
Низкие уровни отражают общее истощение антиоксидативной системы организма / Low levels reflect general depletion of the body's antioxidant system |
[94] |
|
Продукты окисления липидов (малоновый диальдегид, 4-гидроксиноненаль) / Lipid oxidation products (malonic dialdehyde, 4-hydroxynonenal) |
Отражают степень деградации нейрональных мембран с повышением транмембранного транспорта возбуждающих аминокислот, ионов Ca²⁺ и K⁺ / Reflect the level of neuronal membranes degradation with increased transmembrane transport of excitatory amino acids, Ca²⁺ and K⁺ ions |
[81] |
|
Продукты окисления ДНК (8-оксо-2'-дезоксигуанозин) и РНК (7,8-дигидрокси-8-оксогуанин) / Oxidation products of DNA (8-oxo-2'-deoxyguanosine) and RNA (7,8-dihydroxy-8-oxoguanine) |
Биомаркер степени оксидативного повреждения нуклеиновых кислот / Biomarker of the level of oxidative damage to nucleic acids |
[95][96] |
|
3-нитротирозин / 3-nitrotirosine |
Стабильный биомаркер оксидативного стресса, отражающий окисление тирозина / Stable biomarker of oxidative stress reflecting tyrosine oxidation |
[97] |
Примечание. АФК – активные формы кислорода; NO (англ. nitric oxide) – оксид азота; НАДФ(Н) – никотинамидадениндинуклеотидфосфат; ДНК – дезоксирибонуклеиновая кислота; РНК – рибонуклеиновая кислота; HMGB1 (англ. high-mobility group protein B1) – белок высокой подвижности 1-й группы; NF-κB (англ. nuclear factor kappa B) – ядерный фактор каппа В.
Note. ROS – reactive oxygen species; NO – nitric oxide; NADP(H) – nicotinamide adenine dinucleotide phosphate; DNA – deoxyribonucleic acid; RNA – ribonucleic acid; HMGB1 – high-mobility group protein B1; NF-κB – nuclear factor kappa B.
МикроРНК являются важными инструментами эпигенетического контроля экспрессии генов и участвуют в регуляции различных физиологических и патологических процессов, играющих существенную роль в механизмах развития МетС и ПЭП-МетС, включая оксидативный стресс [98][99] и системное воспаление [55][100] (табл. 4). Они оказывают значительное влияние на течение ПЭП-МетС, а также ассоциированы с течением самой эпилепсии, поэтому рассматриваются в исследованиях последнего десятилетия в качестве чувствительных и специфичных эпигенетических биомаркервов эпилептогенеза и ПЭП-МетС [52][101–104]. Например, микроРНК способны модулировать синаптическую передачу, регулируя экспрессию и пропускающую способность рецепторов возбуждающих и тормозных нейромедиаторов, тем самым оказывая эпилептогенное и противоэпилептическое воздействие на ЦНС [105].
Таблица 4. Роль циркулирующих микроРНК в механизмах патогенеза метаболического синдрома, индуцированного приемом противоэпилептических препаратов
Table 4. The role of circulating microRNAs in the mechanisms of antiepileptic drug-induced metabolic syndrome pathogenesis
|
Механизм патогенеза / Mechanism of pathogenesis |
Роль микроРНК / Role of microRNAs |
Источники / References |
|
|
Ингибиторы / Inhibitors |
Индукторы / Inductors |
||
|
Системное воспаление / Systemic inflammation |
miR-7, miR-9, miR-10a, miR-15a, miR-16, miR-24, miR-31, miR-124, miR-125, miR-126, miR-142, miR-143, miR-146, miR-149, miR-150, miR-210, miR-223, miR-363 |
miR-21, miR-23a, miR-27a, miR-29a, miR-34a, miR-34c, miR-92a, miR-132, miR-138, miR-155, miR-196, miR-200, let-7a |
[55][100][106] |
|
Оксидативный стресс / Oxidative stress |
miR-19b, miR-20a, miR-24, miR-99a, miR-125b, miR-128-3p, miR-141, miR-146, miR-152, miR-200a, miR-200c, miR-210, miR-221, miR-455, miR-601, miR-626 |
miR-1, miR-21, miR-23b, miR-27a, miR-28, miR-29, miR-34a, miR-92a, miR-93, miR-101, miR-106b, miR-128, miR-129, miR-140, miR-142, miR-144, miR-148, miR-153, miR-155, miR-181c, miR-193b, miR-320, miR-365, miR-375, miR-383, miR-495, miR-503, miR-802 |
[98][99] |
Примечание. РНК – рибонуклеиновая кислота.
Note. RNA – ribonucleic acid.
МикроРНК как биомаркеры системного воспаления
МикроРНК способны влиять на различные стадии системного воспалительного ответа и регулируют как положительную, так и отрицательную обратную связь с развитием ПЭП-МетС, в т.ч. экспрессию провоспалительных цитокинов, ограничивая инвазию патогенов, факторов транскрипции, активацию репарации повреждения нейронов и нейроглии, регуляцию активации иммунокомпетентных клеток для поддержания тканевого гемостаза (см. табл. 4) [55][59].
Нами обобщены ранее изученные микроРНК-ингибиторы системных воспалительных реакций (miR-7, miR-9, miR-10a, miR-15a, miR-16, miR-24, miR-31, miR-124, miR-125, miR-126, miR-142, miR-143, miR-146, miR-149, miR-150, miR-210, miR-223, miR-363), регулирующие синтез, процессинг биомаркеров системного и локального воспаления и их факторов транскрипции [55][100].
Противовоспалительные механизмы этих микроРНК разнообразны и связаны с модуляцией факторов транскрипции и медиаторов воспаления. Так, miR-7 подавляет ангиогенез и утолщение гладкой мускулатуры церебральных сосудов в поврежденных тканях головного мозга, ингибируя фактор роста эндотелия сосудов (англ. vascular endothelial growth factor, VEGF), а miR-9 активирует путь янус-киназы и активатора транскрипции JAK1/STAT61, препятствуя образованию инфламмасом в макрофагах и развитию глиоза в ЦНС [100][107].
Как описано выше, NF-κB индуцирует и поддерживает системное воспаление, а также является одним из ключевых звеньев эпилептогенеза и ПЭП-МетС, повышая синтез провоспалительных молекул – ИЛ-6, ИЛ-8, моноцитарного хемотаксического протеина 1 (англ. monocyte chemoattractant protein 1, MCP-1) и васкулярной молекулы клеточной адгезии 1 (англ. vascular cell adhesion molecule 1, VCAM-1) [4][58][108][109]. Одним из центральных регуляторов NF-κB является miR-10a, ингибрующая экспрессию MAPK и белка, содержащего повторы β-трансдуцина (англ. β-transducin repeat-containing protein, β-TrCP), которые активируют NF-κB. По схожему механизму, подавляя сигналы MAPK, инактивируют пути TLR4/MyD88/NF-κB другие микроРНК (miR-125b, miR-143 и miR-146 [100].
Другим противовоспалительным механизмом обладает miR-124, ограничивающая секрецию провоспалительных цитокинов (ИЛ-6 и ФНО-α) макрофагами путем ингибирования экспрессии ФНО-α-превращающего фермента [100].
МикроРНК miR-126 уменьшает сосудистое воспаление, ингибируя белки эндотелия, усиливающие ангиогенез (SPRED1, фосфоинозитолкиназа-3, VCAM1) [110].
МикроРНК miR-142 ингибирует ИЛ-1-рецептор-ассоциированную киназу (англ. interleukin-1 receptor-associated kinase 1, IRAK1) и инактивирует NF-κB, снижая тем самым продукцию провоспалительных цитокинов ФНО-α и ИЛ-6 [100].
Некоторые микроРНК могут непосредственно регулировать экспрессию провоспалительных цитокинов. Например, гиперэкспрессия miR-181a подавляет синтез ФНО-α, а гипоэкспрессия miR-146a приводит к повышению уровней ИЛ-1β, ИЛ-18 [100].
Препятствуя передаче сигналов на TLR4, miR-223 способна инактивировать M1-макрофаги, тем самым выключая одно из звеньев системного воспалительного ответа [55][100][111].
МикроРНК-индукторы системного воспалительного ответа (miR-21, miR-23a, miR-27a, miR-29a, miR-34a, miR-34c, miR-92a, miR-132, miR-138, miR-155, miR-200, miR-let-7a), наоборот, ассоциированы с активацией факторов транскрипции медиаторов воспаления [55][100]. Например, miR-23a, инициируя провоспалительный путь NF-κB в активированных макрофагах, одновременно с этим подавляет ось JAK1/STAT6, которая препятствует образованию инфламмасом. Еще одним активатором пути NF-κB является miR-let-7a, которая подавляет экспрессию ингибитора κB-киназы [100][107]. МикроРНК miR-21 усиливает повреждение нейронов, запуская процессы неоангиогенеза в воспаленных тканях головного мозга путем активации VEGF-A. Гиперэкспрессия miR-21 в адипоцитах у пациентов с ПЭП-МетС усиливает их дифференцировку, поддерживает вялотекущее локальное воспаление, повышая риск развития центрального ожирения [100][112][113]. Сигнальный путь NF-κB, а также пути MyD88 и TRIF активируются при гиперэкспрессии miR-138 и miR-155, приводя к повышению уровней провоспалительных медиаторов, например HMGB1, ИЛ-6 [60][100][114].
Паралоги семейства miR-34 (miR-34a и miR-34c) инактивируют G-белки, способствуя беспрепятственному высвобождению провоспалительных цитокинов и хемокинов в активированных клетках, запуская и усугубляя системный воспалительный ответ [98][115].
Гиперэкспрессия miR-92a индуцирует экспрессию генов, кодирующих провоспалительные цитокины в макрофагах [98], а гиперэкспрессия miR-196 и miR-200 ассоциирована с повышением синтеза белка Zeb-1, активирующего ЦОГ-2 и MCP-1 в гладкомышечных клетках сосудов [100]. Влияние оси Zeb-1/ЦОГ-2/нейровоспаление на эпилептогенез и ПЭП-МетС рассмотрено нами ранее в разделе, посвященном системному воспалению [7][70][106][116].
МикроРНК как биомаркеры оксидативного стресса
Подобно вариабельному активирующему и ингибирующему влиянию на механизмы реализации системных воспалительных реакций, микроРНК также регулирируют сигнальные пути и ферменты, опосредующие прооксидантное повреждение или антиоксидативные механизмы защиты (см. табл. 4). Ряд микроРНК (miR-19b, miR-20a, miR-24, miR-99a, miR-125b, miR-128-3p, miR-141, miR-146, miR-152, miR-200a, miR-200c, miR-210, miR-221, miR-455, miR-601, miR-626), ассоциированных с цитопротекцией и снижением маркеров оксидативного стресса, модулирует сигнальный путь 2-го типа ядерного фактора, связанного с эритропоэтином (англ. nuclear factor-erythroid 2 related factor, Nrf2), который активируется в ответ на окислительные и электрофильные нарушения. Механизмами, по которым микроРНК регулируют Nrf2, являются изменение ядерной транслокации Nrf2, контроль экспрессии Nrf2 и индуцируемых им медиаторов, регуляция келч-подобного белка Keap1 [98][99][117][118]. Keap1 представляет собой важный регулятор активации Nrf2, необходимый для поддержания нормального (физиологического) уровня АФК путем убиквитинирования и деградации транскрипционного фактора Nrf2 [117].
К антиоксидативным микроРНК, которые ингибируют Keap1, высвобождая из-под его влияния Nrf2, относятся miR-24, miR-152, miR-200a и miR-626 [99]. Окислительный стресс, вызванный приемом ВК, может объясняться снижением активности системы Nrf2 и активацией промотора Keap1. Исходя из этих данных, высказана гипотеза об ожидаемой гиперэкспресии этих микроРНК при терапии вальпроатами, что делает их перспективными эпигенетическими биомаркерами оксидативного стресса при ВК-индуцированном МетС [118].
Регуляция фактора транксрипции Nrf2 с участием микроРНК осуществляется и по другому сценарию с использованием указанных выше регуляторов. Например, гиперэкспрессия miR-125b активирует Nrf2 путем регуляции пероксиредоксина типа L2A (ген PRXL2A) [99], а miR-455 – через регуляцию ферментативной активности гистондеацетилазы 2-го типа (англ. histone deacetylase 2, HDAC2) [98].
МикроРНК также модулируют экспрессию антиоксидативных ферментов (SIRT, НAД(Ф)H-куинондегидрогеназа 1 (NQO1) и гемоксидаза (HO-1)). Сиртуины – это НАД⁺-зависимые деацетилазы, являющиеся важными антиоксидантными ферментами, активирующими Nrf2 как напрямую, так и через описанные выше механизмы. SIRT1 является наиболее изученным ферментом при ПЭП-МетС. В частности, длительный прием ВК приводит к снижению плазменного уровня SIRT1, ассоциированного с развитием ВК-индуцировнного МетС [99][119]. SIRT3 активирует протеины forkhead box O1 и О2 в митохондриях и регулирует митохондриальный метаболизм, защищая их от оксидативного повреждения при нейровоспалении и эпилептогенезе [120][121]. В частности, miR-128-3p повышает уровни SIRT1, SIRT3, NQO1 и HO-1, а также активирует Nrf2. В то же время miR-141 уменьшает оксидативный стресс и его последствия через систему Nrf2/Keap1, повышает уровень HO-1, подавляет миграцию и пролиферацию гладкомышечных клеток сосудов, что препятствует развитию ПЭП-МетС в целом и атерогенеза в частности [98].
MiR-455, помимо активации системы Nrf2, разрушает белок куллин-3, который убиквитирует связанные с антиоксидативными ферментами гены (NQO1, HO-1 и GCLC) [98].
Дополнительным антиоксидантным механизмом микроРНК является ингибирование прооксидативных ферментов (например, NOX4, экспрессия которой подавляется miR-99a и miR-146 напрямую), тем самым снижая уровни АФК и предотвращая развитие оксидативного стресса [99][122].
Ряд микроРНК (miR-1, miR-21, mir-23b, miR-27a, miR-28, miR-29, miR-34a, miR-92a, miR-93, miR-101, miR-106b, miR-128, miR-129-3p, miR-140, miR-142, miR-144, miR-148, miR-153, miR-155, miR-181c, miR-193b, miR-320, miR-365, miR-375, miR-383, miR-495, miR-503, miR-802) способны индуцировать оксидативный стресс, подавляя активность фактора транскрипции Nrf2 или ингибируя антиоксидативные SIRT-белки [98][99].
Эти механизмы реализации оксидативного стресса осуществляются с участем miR-23b и miR-128, а другие микроРНК (miR-27а, miR-34a, miR-142, miR-144 и miR-153) подавляют только Nrf2, снижая экспрессию его гена NRF2 (miR-34a) или ингибируя его транскрипцию (miR-27а, miR-34a, miR-142, miR-144, miR-153) [98][99].
На активность антиоксидативных ферментов влияют следующие микроРНК: miR-92a (ингибирует SIRT1, активирует синтазу NO и инфламмасомы в М1-моноцитах) [99], miR-140 (ингибирует SIRT2, HO-1, NQO1 и forkhead box O3a, а также индуцирует экспрессию Keap1) [96], miR-320 (ингибирует Nrf2 и нижестоящий HO-1) [98], miR-383 (ингибирует пероксиредоксин 3-го типа) [99].
Проблема МетС и ПЭП-МетС активно обсуждается и изучается в последние годы, что привело к динамическому пересмотру клинических и лабораторных биомаркеров и расширило наше понимание механизмов их патогенеза. Кандидаты в биомаркеры МетС и ПЭП-МетС отбираются в результате многочисленных доклинических и клинических исследований из большого числа молекул, вырабатываемых клетками и тканями экспериментальных животных и человека.
Эпигенетические биомаркеры обладают рядом преимуществ по сравнению с ранее обнаруженными клиническими и лабораторными (генетическими, биохимическими, гормональными) биомаркерами, поскольку могут предоставить врачу важную информацию о текущей функции генов в отдельных типах клеток. Кроме того, они отражают информацию о влиянии модифицируемых факторов окружающей среды и образа жизни пациента с эпилепсией, приема ПЭП и других ЛС, назначаемых по поводу сопутствующих заболеваний, на механизмы развития метаболических расстройств. Преимуществом микроРНК как эпигенетических биомаркеров ПЭП-МетС являются [123]:
– возможность предоставления информации о естественном или отягощенном течении эпилепсии;
– высокая стабильность в биообразцах (плазме, слюне, моче, сперме, вагинальном секрете, грудном молоке, ликворе);
– высокая стабильность в основных типах тканевых препараторов (свежих и замороженных тканях, пятнах засохшей крови (карты Гатри), парафиновых блоках, фиксированных формалином).
Таким образом, микроРНК идеально подходят на роль перспективных эпигенетических биомаркеров ПЭП-МетС [124][125] для оценки безопасности применения ПЭП и оценки их терапевтической резистентности [126][127].
Внедрение платформ для генетического секвенирования нового поколения, например Illumina (Illumina Inc., США), и их применение для секвенирования микроРНК, параллельная разработка сложных вычислительных методов для анализа микроРНК в будущем позволят расширить возможности лабораторной диагностики с использованием эпигенетических биомаркеров для прогнозирования и ранней диагностики ПЭП-МетС у пациентов с эпилепсией [128][129]. Однако следует признать, что количество микроРНК, получивших широкое клиническое признание и нуждающихся во внедрении в реальную клиническую практику, пока невелико. Рекомендации по использованию микроРНК, одобренные Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (англ. U.S. Food and Drug Administration, FDA), Европейским агентством лекарственных средств (англ. European Medicines Agency, EMA), Росздравнадзором и Минпромторгом России, пока носят одиночный характер.
Подготовленный нами обзор позволил систематизировать ранее изученные микроРНК, которые участвуют в регуляции системного воспаления и усиливающего его оксидативного стресса (см. табл. 4), и обобщить их роль как прогностических и предиктивных биомаркеров системного воспаления и оксидативного стресса (одних из ведущих патогенетическх механизмов ПЭП-МетС), что представляет несомненный клинический интерес в неврологии (рис. 1).
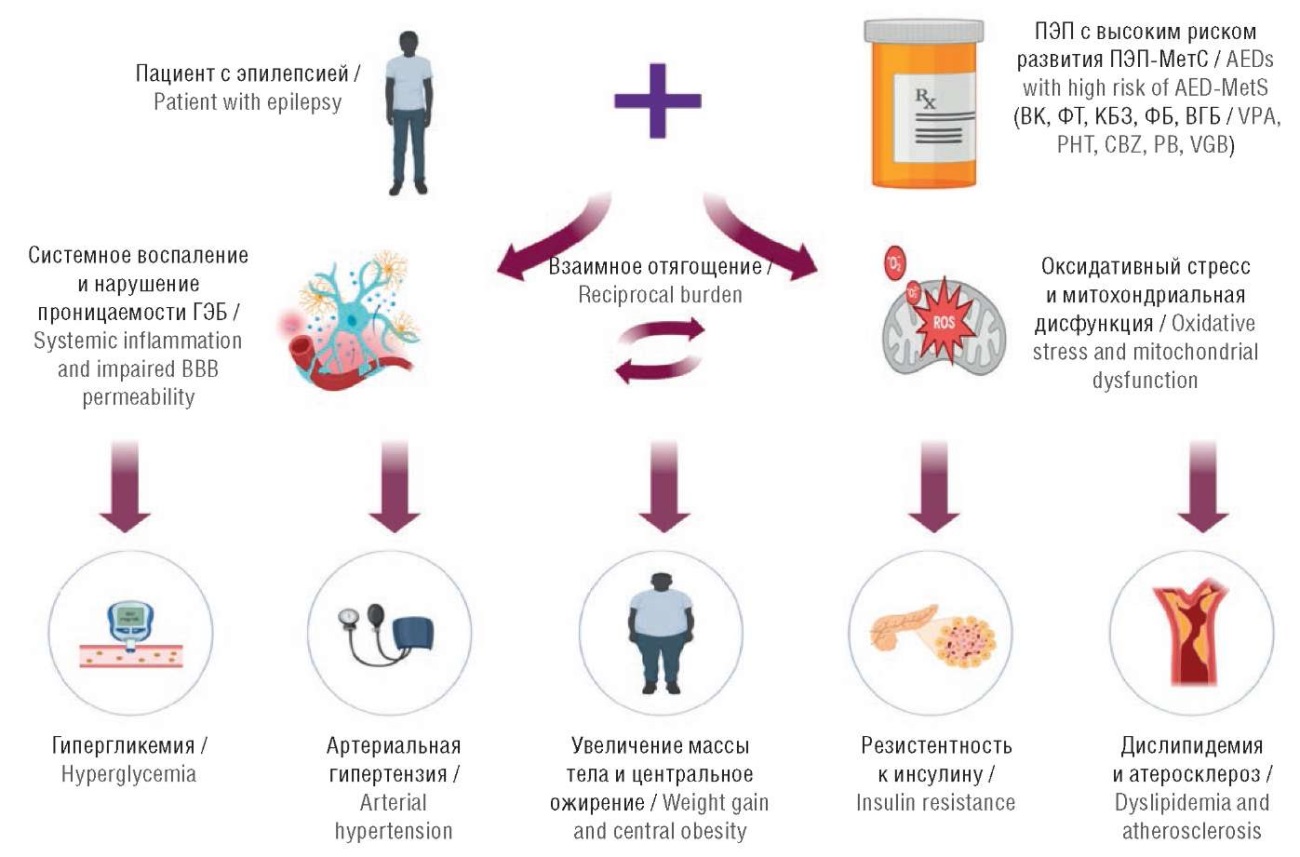
Рисунок 1. Взаимоотягощающий эффект системного воспаления и оксидативного стресса, индуцированных длительным приемом противоэпилептических препаратов, на развитие метаболического синдрома у пациентов с эпилепсией (рисунок подготовлен авторами).
ГЭБ – гематоэнцефалический барьер; ПЭП – противоэпилептический препарат; ПЭП-МетС – метаболический синдром, индуцированный приемом противоэпилептических препаратов; ВК – вальпроевая кислота; ФТ – фенитоин; КБЗ – карбамазепин; ФБ – фенобарбитал; ВГБ – вигабатрин
Figure 1. The reciprocal burden effect of systemic inflammation and oxidative stress induced by prolonged use of antiepileptic drugs on developing metabolic syndrome in patients with epilepsy (the figure was created by the authors).
BBB – blood-brain barrier; AED – antiepileptic drug; AED-MetS – metabolic syndrome induced by antiepileptic drugs; VPA – valproic acid; PHT – phenytoin; CBZ – carbamazepine; PB – phenobarbital; VGB – vigabatrin
Следует признать, что пока большинство исследований микроРНК при рассматриваемой патологии являются доклиническими и выполены на животных моделях и клеточных линиях, а клинические исследования с участием пациентов с МетС и ПЭП-МетС пока единичны и охватывают небольшие выборки. Это является ограничением настоящего обзора и не позволяет сформулировать убедительные обобщающие выводы и рекомендации к применению изученных эпигенетических биомаркеров в реальной клинической практике эпилептолога.
На основании полученных результатов было решено разделить рассматриваемые микроРНК в зависимости от риска развития системного воспалительного ответа и оксидативного стресса на три группы: низкий, средний и высокий риск (рис. 2). К группе низкого риска мы отнесли микроРНК с протективными эффектами как на окислительный стресс, так и на системное воспаление. Группа среднего риска обладает и протективными, и предиктивными свойствами. Группа высокого риска имеет сугубо предиктивные свойства в отношении системного воспаления, оксидативного стресса и, как следствие, МетС.
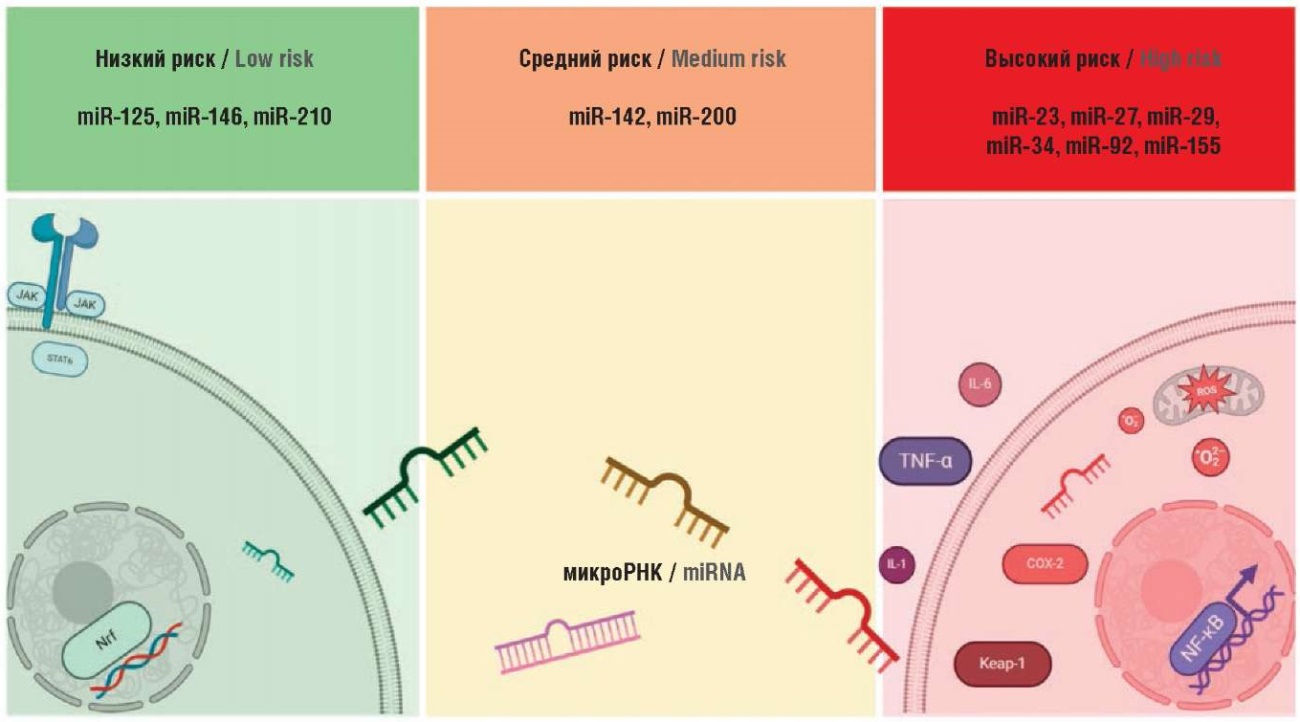
Рисунок 2. Градация риска развития метаболического синдрома, индуцированного приемом противоэпилептических препаратов, в зависимости от индукции системного воспалительного ответа и оксидативного стресса (рисунок подготовлен авторами).
miR – микроРНК; JAK (англ. janus kinase) – янус-киназа; ; STAT6 (англ. signal transducer and activator of transcription 6) – сигнальный преобразователь и активатор транскрипции 6; Nrf2 (англ. nuclear factor-erythroid 2 related factor) – ядерный фактор, связанный с эритропоэтином; IL (англ. interleukin) – интерлейкин; ROS (англ. reactive oxygen species) – активные формы кислорода; TNF-α (англ. tumor necrosis factor alpha) – фактор некроза опухоли альфа; COX-2 (англ. cyclooxygenase-2) – циклооксигеназа-2; Keap-1 (англ. kelch-like ECH-associated protein 1) – келч-подобный ECH-ассоциированный белок 1; NF-κB (англ. nuclear factor kappa B) – ядерный фактор каппа B
Figure 2. Risk-grading system for developing metabolic syndrome related to antiepileptic drugs, depending on induction of systemic inflammatory response and oxidative stress (the figure was created by the authors).
miR – microRNA; JAK – janus kinase; STAT6 – signal transducer and activator of transcription 6; Nrf2 – nuclear factor-erythroid 2 related factor; IL – interleukin; ROS – reactive oxygen species; TNF-α – tumor necrosis factor alpha; COX-2 – cyclooxygenase-2; Keap-1 – kelch-like ECH-associated protein 1; NF-κB – nuclear factor kappa B
К наиболее перспективным с позиции одновременного индуцирующего влияния на системное воспаление и оксидативный стресс микроРНК можно отнести miR-27a, которая ингибирует транскрипцию антиоксидативного фактора Nrf2 [98] и запускает транскрипцию провоспалительного NF-κB [100]. Напротив, miR-142 демонстрирует одновременно противовоспалительные и прооксидативные эффекты, ингибируя IRAK1 и инактивируя NF-κB [100], при этом она подавляет транскрипцию Nrf2 [98].
К протективным микроРНК в отношении как оксидативного стресса, так и системного воспаления можно отнести miR-146 и miR-210. Антиоксидативные свойства miR-146 обусловлены подавлением активности НAД(Ф)H-оксидазы [99], а противовоспалительный эффект – инактивацией пути TLR4/MyD88/NF-κB через ингибирование MAPK [100]. МикроРНК miR-210 реализует свой противовоспалительный потенциал, непосредственно ингибируя NF-κB [100], а ее антиоксидативные эффекты обусловлены снижением образования АФК [98].
Проблема прогнозирования и ранней диагностики ПЭП-МетС актуальна для современной эпилептологии, но далека от разрешения. Перспективным направлением ее решения является поиск предиктивных эпигенетических биомаркеров и разработка панелей (сигнатур) на основе циркулирующих микроРНК, изменение экспрессии которых ассоциировано с высоким риском рассматриваемой НР фармакотерапии эпилепсии.
С позиции персонализированной медицины эпигеномика представляет собой новый этап не только изучения эфективности ПЭП, но и прогнозирования развития НР со стороны эндокринной и других систем. Это объясняет важность планирования и проведения клинических исследований чувствительности и специфичности сигнатур циркулирующих микроРНК в крови больных эпилепсией с ПЭП-МетС и без него.
Расширение наших знаний о чувствительных и специфичных предиктивных биомаркерах ПЭП-МетС позволит повысить безопасность лечения эпилепсии и комплаентность пациентов к соблюдению дозы и режима приема ПЭП.
1. JAK1 (англ. janus kinase 1) – янус-киназа 1; STAT6 (англ. signal transducer and activator of transcription 6) – сигнальный белок и активатор транскрипции 6.
1. Epilepsy: a public health imperative. Available at: https://www.who.int/publications/i/item/epilepsy-a-public-health-imperative (accessed 24.01.2025)
2. Zhang Y.J., Kong X.M., Lv J.J., et al. Analysis of the global burden of disease study highlights the global, regional, and national trends of idiopathic epilepsy epidemiology from 1990 to 2019. Prev Med Rep. 2023; 36: 102522. https://doi.org/10.1016/j.pmedr.2023.102522.
3. Pitkänen A., Lukasiuk K., Dudek F.E., Staley K.J. Epileptogenesis. Cold Spring Harb Perspect Med. 2015; 5 (10): a022822. https://doi.org/10.1101/cshperspect.a022822.
4. Meng F., Yao L. The role of inflammation in epileptogenesis. Acta Epileptologica. 2020; 2 (1): 15. https://doi.org/10.1186/s42494-020-00024-y.
5. Borowicz-Reutt K.K., Czuczwar S.J. Role of oxidative stress in epileptogenesis and potential implications for therapy. Pharmacol Rep. 2020; 72 (5): 1218–26. https://doi.org/10.1007/s43440-020-00143-w.
6. Xie J., Van Hoecke L., Vandenbroucke R.E. The impact of systemic inflammation on Alzheimer's disease pathology. Front Immunol. 2022; 12: 796867. https://doi.org/10.3389/fimmu.2021.796867.
7. Shimada T., Takemiya T., Sugiura H., Yamagata K. Role of inflammatory mediators in the pathogenesis of epilepsy. Mediators Inflamm. 2014; 2014: 901902. https://doi.org/10.1155/2014/901902.
8. Zhao Y., Shao W., Zhu Q., et al. Association between systemic immuneinflammation index and metabolic syndrome and its components: results from the National Health and Nutrition Examination Survey 2011–2016. J Transl Med. 2023; 21 (1): 691. https://doi.org/10.1186/s12967-023-04491-y.
9. Ramos-González E.J., Bitzer-Quintero O.K., Ortiz G., et al. Relationship between inflammation and oxidative stress and its effect on multiple sclerosis. Neurologia (Engl Ed). 2024; 39 (3): 292–301. https://doi.org/10.1016/j.nrleng.2021.10.010.
10. Magheru C., Magheru S., Coltau M., et al. Antiepileptic drugs and their dual mechanism of action on carbonic anhydrase. J Clin Med. 2022; 11 (1): 2614. https://doi.org/10.3390/jcm11092614.
11. Biso L., Aringhieri S., Carli M., et al. Therapeutic drug monitoring in psychiatry: enhancing treatment precision and patient outcomes. Pharmaceuticals. 2024; 17: 642. https://doi.org/10.3390/ph17050642.
12. Costa B., Vale N. Understanding lamotrigine’s role in the CNS and possible future evolution. Int J Mol Sci. 2023; 24 (7): 6050. https://doi.org/10.3390/ijms24076050.
13. Jakovljević D., Nikolić M., Jovanović V., et al. Influence of longterm anti-seizure medications on redox parameters in human blood. Pharmaceuticals. 2024; 17 (1): 130. https://doi.org/10.3390/ph17010130.
14. Kaushik S., Chopra D., Sharma S., Aneja S. Adverse drug reactions of anti-epileptic drugs in children with epilepsy: a cross-sectional study. Curr Drug Saf. 2019; 14: 217–24. https://doi.org/10.2174/1574886314666190311112710.
15. Nazish S. Obesity and metabolic syndrome in patients with epilepsy, their relation with epilepsy control. Ann Afr Med. 2023; 22 (2): 136–44. https://doi.org/10.4103/aam.aam_139_22.
16. Beyene Kassaw A., Tezera Endale H., Hunie Tesfa K., Derbew Molla M. Metabolic syndrome and its associated factors among epileptic patients at Dessie Comprehensive Specialized Hospital, Northeast Ethiopia; a hospital-based comparative cross-sectional study. PLoS One. 2022; 17 (12): e0279580. https://doi.org/10.1371/journal.pone.0279580.
17. Nair S.S., Harikrishnan S., Sarma P.S., Thomas S.V. Metabolic syndrome in young adults with epilepsy. Seizure. 2016; 37: 61–4. https://doi.org/10.1016/j.seizure.2016.03.0021-2.
18. Chen B., Choi H., Hirsch L.J., et al. Cosmetic side effects of antiepileptic drugs in adults with epilepsy. Epilepsy Behav. 2015; 42: 129–37. https://doi.org/10.1016/j.yebeh.2014.10.021.
19. Shnayder N.A., Grechkina V.V., Trefilova V.V., et al. Valproate-induced metabolic syndrome. Biomedicines. 2023; 11 (5): 1499. https://doi.org/10.3390/biomedicines11051499.
20. Tien N., Wu T.Y., Lin C.L., et al. Association of epilepsy, anti-epileptic drugs (AEDs), and type 2 diabetes mellitus (T2DM): a populationbased cohort retrospective study, impact of AEDs on T2DM-related molecular pathway, and via peroxisome proliferator-activated receptor γ transactivation. Front Endocrinol. 2023; 14: 1156952. https://doi.org/10.3389/fendo.2023.1156952.
21. Rakitin A., Kõks S., Haldre S. Metabolic syndrome and anticonvulsants: a comparative study of valproic acid and carbamazepine. Seizure. 2016; 38: 11–6. https://doi.org/10.1016/j.seizure.2016.03.008.
22. Kovac S., Dinkova Kostova A.T., Herrmann A.M., et al. Metabolic and homeostatic changes in seizures and acquired epilepsy-mitochondria, calcium dynamics and reactive oxygen species. Int J Mol Sci. 2017; 18 (9): 1935. https://doi.org/10.3390/ijms18091935.
23. Meenakshi-Sundaram S., Sankaranarayanan M. Epilepsy, phenytoin, and atherogenic risk – current perspectives. Neurology India. 2021; 69 (4): 962–3. https://doi.org/10.4103/0028-3886.325320.
24. Asranna A., Thomas S.V. Metabolic effects of anti-seizure medications: a time to reevaluate risks? Neurol India. 2021; 69 (4): 964–5. https://doi.org/10.4103/0028-3886.325336.
25. Li Y.X., Guo W., Chen R.X., et al. The relationships between obesity and epilepsy: a systematic review with meta-analysis. PLoS One. 2024; 19 (8): e0306175. https://doi.org/10.1371/journal.pone.0306175.
26. Beghi E., Shorvon S. Antiepileptic drugs and the immune system. Epilepsia. 2011; 52 (3): 40–4. https://doi.org/10.1111/j.1528-1167.2011.03035.x.
27. Martinc B., Grabnar I., Milosheska D., et al. A cross-sectional study comparing oxidative stress in patients with epilepsy treated with old and new generation antiseizure medications. Medicina. 2024; 60 (8): 1299. https://doi.org/10.3390/medicina60081299.
28. Шнайдер Н.А., Гречкина В.В., Киссин М.Я. и др. Роль нейропептида Y в развитии вальпроат-индуцированного расстройства пищевого поведения. Эпилепсия и пароксизмальные состояния. 2024; 16 (4): 349–61. https://doi.org/10.17749/2077-8333/epi.par.con.2024.207.
29. Dehury S., Patro P., Sahu L., et al. Evaluation of metabolic parameters on use of newer antiepileptics versus conventional antiepileptics in patients of generalised tonic-clonic seizure: an observational study. Cureus. 2023; 15 (2): e35181. https://doi.org/10.7759/cureus.35181.
30. Kośmider K., Kamieniak M., Czuczwar S.J., Miziak B. Second generation of antiepileptic drugs and oxidative stress. Int J Mol Sci. 2023; 24 (4): 3873. https://doi.org/10.3390/ijms24043873.
31. Swathi B., Aruna D. Evaluation of antioxidant effects of antiepileptic drugs in adult epileptic patients: an open label, non randomised interventional study. J Сlinical Diagn Res. 2022; 16 (10): 10–4. https://doi.org/10.7860/JCDR/2022/57376.17106.
32. Sheth R.D., Montouris G. Metabolic effects of AEDs: impact on body weight, lipids and glucose metabolism. Int Rev Neurobiol. 2008; 83: 329–46. https://doi.org/.1016/S0074-7742(08)00019-6.
33. Stols-Gonçalves D., Tristão L.S., Henneman P., Nieuwdorp M. Epigenetic markers and microbiota/metabolite-induced epigenetic modifications in the pathogenesis of obesity, metabolic syndrome, type 2 diabetes, and non-alcoholic fatty liver disease. Curr Diab Rep. 2019; 19 (6): 31. https://doi.org/10.1007/s11892-019-1151-4.
34. Pant R., Firmal P., Shah V.K., et al. Epigenetic regulation of adipogenesis in development of metabolic syndrome. Front Cell Dev Biol. 2021; 8: 619888. https://doi.org/10.3389/fcell.2020.619888.
35. Ramirez K., Niraula A., Sheridan J.F. GABAergic modulation with classical benzodiazepines prevent stress-induced neuro-immune dysregulation and behavioral alterations. Brain Behav Immun. 2016; 51: 154–68. https://doi.org/10.1016/j.bbi.2015.08.011.
36. Gramaglia E., Ramella Gigliardi V., Olivetti I., et al. Impact of shortterm treatment with benzodiazepines and imidazopyridines on glucose metabolism in healthy subjects. J Endocrinol Invest. 2014; 37 (2): 203–6. https://doi.org/10.1007/s40618-013-0016-y.
37. Bekkouche L., Bouchenak M., Malaisse W.J., Yahia D.A. The Mediterranean diet adoption improves metabolic, oxidative, and inflammatory abnormalities in Algerian metabolic syndrome patients. Horm Metab Res. 2014; 46 (4): 274–82. https://doi.org/10.1055/s-0033-1363657.
38. Biomarkers definitions working group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001; 69 (3): 89–95. https://doi.org/10.1067/mcp.2001.113989.
39. Califf R.M. Biomarker definitions and their applications. Exp Biol Med. 2018; 243 (3): 213–21. https://doi.org/10.1177/1535370217750088.
40. Шнайдер Н.А., Гречкина В.В., Петрова М.М., Насырова Р.Ф. Клиническая картина вальпроат-индуцированного метаболического синдрома. Забайкальский медицинский вестник. 2023; 3: 89–105. https://doi.org/10.52485/19986173_2023_3_89.
41. Srikanthan K., Feyh A., Visweshwar H., et al. Systematic review of metabolic syndrome biomarkers: a panel for early detection, management, and risk stratification in the west virginian population. Int J Med Sci. 2016; 13 (1): 25–38. https://doi.org/10.7150/ijms.13800.
42. Cho Y., Lee S.Y. Useful biomarkers of metabolic syndrome. Int J Environ Res Public Health. 2022; 19 (22): 15003. https://doi.org/10.3390/ijerph192215003.
43. Lauschke V.M., Zhou Y., Ingelman-Sundberg M. Novel genetic and epigenetic factors of importance for inter-individual differences in drug disposition, response and toxicity. Pharmacol Ther. 2019; 197: 122–52. https://doi.org/10.1016/j.pharmthera.2019.01.002.
44. Reynolds E.H. Antiepileptic drugs, folate one-carbon metabolism, genetics, and epigenetics: congenital, developmental, and neuropsychological risks and antiepileptic action. Epilepsia. 2024; 65 (12): 3469–73. https://doi.org/10.1111/epi.18120.
45. Soler-Botija C., Gálvez-Montón C., Bayés-Genís A. Epigenetic biomarkers in cardiovascular diseases. Front Genet. 2019; 10: 950. https://doi.org/10.3389/fgene.2019.00950.
46. Kumar S., Shanker O.R., Banerjee J., et al. Epigenetics in epilepsy. Prog Mol Biol Transl Sci. 2023; 198: 249–69. https://doi.org/10.1016/bs.pmbts.2023.01.005.
47. Миронова О.Ю., Бердышева М.В., Елфимова Е.М. МикроРНК: взгляд клинициста на состояние проблемы. Часть 2. МикроРНК в качестве биомаркера. Евразийский кардиологический журнал. 2023; 2: 64–71. https://doi.org/10.38109/2225-1685-2023-2-64-71.
48. Dexheimer P.J., Cochella L. MicroRNAs: from mechanism to organism. Front Cell Dev Biol. 2020; 8: 409. https://www.doi.org/10.3389/fcell.2020.00409.
49. Pozniak T., Shcharbin D., Bryszewska M. Circulating microRNAs in medicine. Int J Mol Sci. 2022; 23 (7): 3996. https://www.doi.org/10.3390/ijms23073996.
50. Brandão-Lima P.N., de Carvalho G.B., Payolla T.B., et al. Circulating microRNAs showed specific responses according to metabolic syndrome components and sex of adults from a population-based study. Metabolites. 2022; 13 (1): 2. https://www.doi.org/10.3390/metabo13010002.
51. Solís-Toro D., Mosquera Escudero M., García-Perdomo H.A. Association between circulating microRNAs and the metabolic syndrome in adult populations: a systematic review. Diabetes Metab Syndr. 2022; 16 (1): 102376. https://www.doi.org/10.1016/j.dsx.2021.102376.
52. Ghafouri-Fard S., Hussen B.M., Abak A., et al. Aberrant expression of miRNAs in epilepsy. Mol Biol Rep, 2022; 49: 5057–74. https://doi.org/10.1007/s11033-022-07188-5.
53. O'Brien J., Hayder H., Zayed Y., Peng C. Overview of MicroRNA biogenesis, mechanisms of actions, and circulation. Front Endocrinol. 2018; 9: 402. https://www.doi.org/10.3389/fendo.2018.00402.
54. Page M.J., McKenzie J.E., Bossuyt P.M., et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. BMJ. 2021; 372: n71. https://doi.org/10.1136/bmj.n71.
55. Carvalho G.B., Brandão-Lima P.N., Payolla T.B., et al. Circulating MiRNAs are associated with low-grade systemic inflammation and leptin levels in older adults. Inflammation. 2023; 46 (6): 2132–46. https://www.doi.org/10.1007/s10753-023-01867-6.
56. Menzel A., Samouda H., Dohet F., et al. Common and novel markers for measuring inflammation and oxidative stress ex vivo in research and clinical practice-which to use regarding disease outcomes? Antioxidants. 2021; 10 (3): 414. https://doi.org/10.3390/antiox10030414.
57. Vezzani A., Viviani B. Neuromodulatory properties of inflammatory cytokines and their impact on neuronal excitability. Neuropharmacology. 2015; 96 (Pt A): 70–82. https://doi.org/10.1016/j.neuropharm.2014.10.027.
58. McElroy P.B., Liang L.P., Day B.J., Patel M. Scavenging reactive oxygen species inhibits status epilepticus-induced neuroinflammation. Exp Neurol. 2017; 298 (Pt A): 13–22. https://doi.org/10.1016/j.expneurol.2017.08.009.
59. Chatterjee B., Sarkar M., Bose S., et al. MicroRNAs: key modulators of inflammation-associated diseases. Semin Cell Dev Biol. 2024; 154 (C): 364–73. https://doi.org/10.1016/j.semcdb.2023.01.009.
60. Balosso S., Liu J., Bianchi M.E., Vezzani A. Disulfide-containing high mobility group box-1 promotes N-methyl-D-aspartate receptor function and excitotoxicity by activating toll-like receptor 4-dependent signaling in hippocampal neurons. Antioxid Redox Signal. 2014; 21 (12): 1726–40. https://doi.org/10.1089/ars.2013.5349.
61. Engel T., Alves M., Sheedy C., Henshall D.C. ATPergic signalling during seizures and epilepsy. Neuropharmacology. 2016; 104: 140–53. https://doi.org/10.1016/j.neuropharm.2015.11.001.
62. Huang C., Chi X.S., Li R., et al. Inhibition of P2X7 receptor ameliorates nuclear factor-kappa B mediated neuroinflammation induced by status epilepticus in rat hippocampus. J Mol Neurosci. 2017; 63 (2): 173–84. https://doi.org/10.1007/s12031-017-0968-z.
63. Iori V., Frigerio F., Vezzani A. Modulation of neuronal excitability by immune mediators in epilepsy. Curr Opin Pharmacol. 2016; 26: 118–23. https://doi.org/10.1016/j.coph.2015.11.002.
64. Li T.R., Jia Y.J., Ma C., et al. The role of the microRNA-146a/complement factor H/interleukin-1β-mediated inflammatory loop circuit in the perpetuate inflammation of chronic temporal lobe epilepsy. Dis Model Mech. 2018; 11 (3): dmm031708. https://doi.org/10.1242/dmm.031708.
65. Garcia-Oscos F., Salgado H., Hall S., et al. The stress-induced cytokine interleukin-6 decreases the inhibition/excitation ratio in the rat temporal cortex via trans-signaling. Biol Psychiatry. 2012; 71 (7): 574–82. https://doi.org/10.1016/j.biopsych.2011.11.018.
66. Esquivel-Rendón E., Vargas-Mireles J., Cuevas-Olguín R., et al. Interleukin 6 dependent synaptic plasticity in a social defeatsusceptible prefrontal cortex circuit. Neuroscience. 2019; 414: 280–96. https://doi.org/10.1016/j.neuroscience.2019.07.002.
67. Balosso S., Ravizza T., Aronica E., Vezzani A. The dual role of TNF-α and its receptors in seizures. Exp Neurol. 2013; 247: 267–71. https://doi.org/10.1016/j.expneurol.2013.05.010.
68. Rustenhoven J., Aalderink M., Scotter E.L., et al. TGF-beta1 regulates human brain pericyte inflammatory processes involved in neurovasculature function. J Neuroinflammation. 2016; 13: 37. https://doi.org/10.1186/s12974-016-0503-0.
69. Musto A.E., Samii M. Platelet-activating factor receptor antagonism targets neuroinflammation in experimental epilepsy. Epilepsia. 2011; 52 (3): 551–61. https://doi.org/10.1111/j.1528-1167.2010.02920.x.
70. Rojas A., Jiang J., Ganesh T., et al. Cyclooxygenase-2 in epilepsy. Epilepsia. 2014; 55 (1): 17–25. https://doi.org/10.1111/epi.12461.
71. Yagami T., Koma H., Yamamoto Y. Pathophysiological roles of cyclooxygenases and prostaglandins in the central nervous system. Mol Neurobiol. 2016; 53 (7): 4754–71. https://doi.org/10.1007/s12035-015-9355-3.
72. Zhong R., Chen Q., Li M., et al. Elevated blood C-reactive protein levels in patients with epilepsy: a systematic review and meta-analysis. Front Neurol. 2019; 10: 974. https://doi.org/10.3389/fneur.2019.00974.
73. Basta-Kaim A., Budziszewska B., Lasoń W. Effects of antiepileptic drugs on immune system. Przegl Lek. 2008; 65 (11): 799–802 (in Polish).
74. Dambach H., Hinkerohe D., Prochnow N., et al. Glia and epilepsy: experimental investigation of antiepileptic drugs in an astroglia/microglia co-culture model of inflammation. Epilepsia. 2014; 55 (1): 184–92. https://doi.org/10.1111/epi.12473.
75. Godhwani N., Bahna S.L. Antiepilepsy drugs and the immune system. Ann Allergy Asthma Immunol. 2016; 117 (6): 634–40. https://doi.org/10.1016/j.anai.2016.09.443.
76. Preiser J.C. Oxidative stress. JPEN J Parenter Enteral Nutr. 2012; 36 (2): 147–54. https://doi.org/10.1177/0148607111434963.
77. Zsurka G., Kunz W.S. Mitochondrial dysfunction and seizures: the neuronal energy crisis. Lancet Neurol. 2015; 14 (9): 956–66. https://doi.org/10.1016/S1474-4422(15)00148-9.
78. Puttachary S., Sharma S., Stark S., Thippeswamy T. Seizure-induced oxidative stress in temporal lobe epilepsy. Biomed Res Int. 2015; 2015: 745613. https://doi.org/10.1155/2015/745613.
79. Kalozoumi G., Kel-Margoulis O., Vafiadaki E., et al. Glial responses during epileptogenesis in Mus musculus point to potential therapeutic targets. PLoS One. 2018; 13 (8): e0201742. https://doi.org/10.1371/journal.pone.0201742.
80. Kamieniak M., Kośmider K., Miziak B., Czuczwar S.J. The oxidative stress in epilepsy – focus on melatonin. Int J Mol Sci. 2024; 25 (23): 12943. https://doi.org/10.3390/ijms252312943.
81. Kobylarek D., Iwanowski P., Lewandowska Z., et al. Advances in the potential biomarkers of epilepsy. Front Neurol. 2019; 10: 685. https://doi.org/10.3389/fneur.2019.00685.
82. Masenga S.K., Kabwe L.S., Chakulya M., Kirabo A. Mechanisms of oxidative stress in metabolic syndrome. Int J Mol Sci. 2023; 24 (9): 7898. https://doi.org/10.3390/ijms24097898.
83. Čolak E., Pap D. The role of oxidative stress in the development of obesity and obesity-related metabolic disorders. J Med Biochem. 2021; 40 (1): 1–9. https://doi.org/10.5937/jomb0-24652.
84. Rotariu D., Babes E.E., Tit D.M., et al. Oxidative stress – complex pathological issues concerning the hallmark of cardiovascular and metabolic disorders. Biomed Pharmacother. 2022; 152: 113238. https://doi.org/10.1016/j.biopha.2022.113238.
85. Touyz R.M., Rios F.J., Alves-Lopes R., et al. Oxidative stress: a unifying paradigm in hypertension. Can J Cardiol. 2020; 36 (5): 659–70. https://doi.org/10.1016/j.cjca.2020.02.081.
86. Marrocco I., Altieri F., Peluso I. Measurement and clinical significance of biomarkers of oxidative stress in humans. Oxid Med Cell Longev. 2017; 2017: 6501046. https://doi.org/10.1155/2017/6501046.
87. Forrester S.J., Kikuchi D.S., Hernandes M.S., et al. Reactive oxygen species in metabolic and inflammatory signaling. Circ Res. 2018; 122 (6): 877–902. https://doi.org/10.1161/CIRCRESAHA.117.311401.
88. Thiruvengadam M., Venkidasamy B., Subramanian U., et al. Bioactive compounds in oxidative stress-mediated diseases: targeting the NRF2/ARE signaling pathway and epigenetic regulation. Antioxidants. 2021; 10 (12): 1859. https://doi.org/10.3390/antiox10121859.
89. Шнайдер Н.А., Гречкина В.В., Архипов В.В., Насырова Р.Ф. Фармакогенетически-информированная фармакометаболомика как инновационный подход к оценке безопасности и риска фармакотерапии препаратами вальпроевой кислоты. Безопасность и риск фармакотерапии. 2023; 11 (4): 450–62. https://doi.org/10.30895/2312-7821-2023-386.
90. Haznedar P., Doğan Ö., Albayrak P., et al. Effects of levetiracetam and valproic acid treatment on liver function tests, plasma free carnitine and lipid peroxidation in childhood epilepsies. Epilepsy Res. 2019; 153: 7–13. https://doi.org/10.1016/j.eplepsyres.2019.03.009.
91. Ignacio-Mejía I., Contreras-García I.J., Mendoza-Torreblanca J.G., et al. Evaluation of the antioxidant activity of levetiracetam in a temporal lobe epilepsy model. Biomedicines. 2023; 11 (3): 848. https://doi.org/10.3390/biomedicines11030848.
92. Lu S.C. Regulation of glutathione synthesis. Mol Aspects Med. 2009; 30 (1–2): 42–59. https://doi.org/10.1016/j.mam.2008.05.005.
93. Kumawat M., Sharma T.K., Singh I., et al. Antioxidant enzymes and lipid peroxidation in type 2 diabetes mellitus patients with and without nephropathy. N Am J Med Sci. 2013; 5 (3): 213–9. https://doi.org/10.4103/1947-2714.109193.
94. Lettieri-Barbato D., Tomei F., Sancini A., et al. Effect of plant foods and beverages on plasma non-enzymatic antioxidant capacity in human subjects: a meta-analysis. Br J Nutr. 2013; 109 (9): 1544–56. https://doi.org/10.1017/S0007114513000263.
95. Li H., Cui S., Wang S., et al. Ultrasensitive UPLC-MS/MS method for analysis of etheno-DNA adducts in human white blood cells. Free Radic Res. 2015; 49 (9): 1049–54. https://doi.org/10.3109/10715762.2015.1006213.
96. Broedbaek K., Siersma V., Henriksen T., et al. Urinary markers of nucleic acid oxidation and cancer in type 2 diabetes. Redox Biol. 2015; 4: 34–9. https://doi.org/10.1016/j.redox.2014.11.010.
97. Meredith S., Parekh G., Towler J., et al. Mapping nitro-tyrosine modifications in fibrinogen by mass spectrometry as a biomarker for inflammatory disease. Free Radic Biol Med. 2014; 75 (1): S50. https://doi.org/10.1016/j.freeradbiomed.2014.10.819.
98. Saha S. Role of microRNA in oxidative stress. Stresses. 2024; 4 (2): 269–81. https://doi.org/10.3390/stresses4020016.
99. Włodarski A., Strycharz J., Wróblewski A., et al. The role of microRNAs in metabolic syndrome-related oxidative stress. Int J Mol Sci. 2020; 21 (18): 6902. https://www.doi.org/10.3390/ijms21186902.
100. Das K., Rao L.V.M. The role of microRNAs in inflammation. Int J Mol Sci. 2022; 23 (24): 15479. https://www.doi.org/10.3390/ijms232415479.
101. Tiwari D., Peariso K., Gross C. MicroRNA-induced silencing in epilepsy: opportunities and challenges for clinical application. Dev Dyn. 2018; 247 (1): 94–110. https://doi.org/10.1002/dvdy.24582.
102. Vasilieva A.A., Timechko E.E., Lysova K.D., et al. MicroRNAs as potential biomarkers of post-traumatic epileptogenesis: a systematic review. Int J Mol Sci. 2023; 24 (20): 15366. https://doi.org/10.3390/ijms242015366.
103. Lysova K.D., Usoltseva A.A., Domoratskaya E.A., et al. miR-134 and miR-106b are circulating biomarkers for temporal lobe epilepsy: pilot study results. Russian Open Medical Journal. 2023; 12: e0303. https://doi.org/10.15275/rusomj.2023.0303.
104. Gottmann P., Ouni M., Zellner L., et al. Polymorphisms in miRNA binding sites involved in metabolic diseases in mice and humans. Sci Rep. 2020; 10: 7202. https://doi.org/10.1038/s41598-020-64326-4.
105. Sonawane S., Všianský V., Brázdil M. MicroRNA-mediated regulation of neurotransmitter receptors in epilepsy: a systematic review. Epilepsy Behav. 2024; 158: 109912. https://doi.org/10.1016/j.yebeh.2024.109912.
106. Porretti J., Dalton G.N., Massillo C., et al. CLCA2 epigenetic regulation by CTBP1, HDACs, ZEB1, EP300 and miR-196b-5p impacts prostate cancer cell adhesion and EMT in metabolic syndrome disease. Int J Cancer. 2018; 143 (4): 897–906. https://doi.org/10.1002/ijc.31379.
107. Sun H., Ma D., Cheng Y., et al. The JAK-STAT signaling pathway in epilepsy. Curr Neuropharmacol. 2023; 21 (10): 2049–69. https://doi.org/10.2174/1570159X21666221214170234.
108. Cai M., Lin W. The function of NF-kappa B during epilepsy, a potential therapeutic target. Front Neurosci. 2022; 16: 851394. https://doi.org/10.3389/fnins.2022.851394.
109. Kracht M., Müller-Ladner U., Schmitz M.L. Mutual regulation of metabolic processes and proinflammatory NF-κB signaling. J Allergy Clin Immunol. 2020; 146 (4): 694–705. https://doi.org/10.1016/j.jaci.2020.07.027.
110. Wang Y., Peng J., Bai S., et al. A PIK3R2 mutation in familial temporal lobe epilepsy as a possible pathogenic variant. Front Genet. 2021; 12: 596709. https://doi.org/10.3389/fgene.2021.596709.
111. Tang X., Chen X., Li X., et al. The TLR4 mediated inflammatory signal pathway might be involved in drug resistance in drug-resistant epileptic rats. J Neuroimmunol. 2022; 365: 577802. https://doi.org/10.1016/j.jneuroim.2021.577802.
112. Rafi S.K., Goering J.P., Olm-Shipman A.J., et al. Anti-epileptic drug topiramate upregulates TGFβ1 and SOX9 expression in primary embryonic palatal mesenchyme cells: implications for teratogenicity. PLoS One. 2021; 16 (2): e0246989. https://doi.org/10.1371/journal.pone.0246989.
113. Castañeda-Cabral J.L., Beas-Zárate C., Rocha-Arrieta L.L., et al. Increased protein expression of VEGF-A, VEGF-B, VEGF-C and their receptors in the temporal neocortex of pharmacoresistant temporal lobe epilepsy patients. J Neuroimmunol. 2019; 328: 68–72. https://doi.org/10.1016/j.jneuroim.2018.12.007.
114. Wang N., Han X., Liu H., et al. Myeloid differentiation factor 88 is upregulated in epileptic brain and contributes to experimental seizures in rats. Exp Neurol. 2017; 295: 23–35. https://doi.org/10.1016/j.expneurol.2017.05.008.
115. Cheng L., Xia F., Li Z., et al. Structure, function and drug discovery of GPCR signaling. Mol Biomed. 2023; 4 (1): 46. https://doi.org/10.1186/s43556-023-00156-w.
116. Poonaki E., Kahlert U.D., Meuth S.G., Gorji A. The role of the ZEB1- neuroinflammation axis in CNS disorders. J Neuroinflammation. 2022; 19 (1): 275. https://doi.org/10.1016/10.1186/s12974-022-02636-2.
117. Ткачев В.О., Меньшикова Е.Б., Зенков Н.К. Механизм работы сигнальной системы NRF2/KEAP1/ARE (обзор). Биохимия. 2011; 76 (4): 502–19.
118. Palsamy P., Bidasee K.R., Shinohara T. Valproic acid suppresses Nrf2/Keap1 dependent antioxidant protection through induction of endoplasmic reticulum stress and Keap1 promoter DNA demethylation in human lens epithelial cells. Exp Eye Res. 2014; 121: 26–34. https://doi.org/10.1016/j.exer.2014.01.021.
119. Zhao M., Li G., Zhao L. The role of SIRT1-FXR signaling pathway in valproic acid induced liver injury: a quantitative targeted metabolomic evaluation in epileptic children. Front Pharmacol. 2024; 15: 1477619. https://doi.org/10.3389/fphar.2024.1477619.
120. Zhang H., Dai S., Yang Y., et al. Role of sirtuin 3 in degenerative diseases of the central nervous system. Biomolecules. 2024; 13 (5): 735. https://doi.org/10.3390/biom13050735.
121. Yang W., Nagasawa K., Munch C., et al. Mitochondrial sirtuin network reveals dynamic sirt3-dependent deacetylation in response to membrane depolarization. Cell. 2016; 167: 985–1000.e21. https://doi.org/10.1016/j.cell.2016.10.016.
122. Tang P., Dang H., Huang J., et al. NADPH oxidase NOX4 is a glycolytic regulator through mROS-HIF1α axis in thyroid carcinomas. Sci Rep. 2018; 8: 15897. https://doi.org/10.1038/s41598-018-34154-8.
123. García-Giménez J.L., Seco-Cervera M., Tollefsbol T.O., et al. Epigenetic biomarkers: current strategies and future challenges for their use in the clinical laboratory. Crit Rev Clin Lab Sci. 2017; 54 (7–8): 529–50. https://doi.org/10.1080/10408363.2017.1410520.
124. Martinez B., Peplow P.V. MicroRNAs as potential biomarkers in temporal lobe epilepsy and mesial temporal lobe epilepsy. Neural Regen Res. 2023; 18 (4): 716–26. https://doi.org/10.4103/1673-5374.354510.
125. Rzepka-Migut B., Paprocka J. Prospects and limitations related to the use of microrna as a biomarker of epilepsy in children: a systematic review. Life. 2021; 11: 26. https://doi.org/10.3390/life11010026.
126. De Benedittis S., Fortunato F., Cava C., et al. Circulating microRNAs as potential novel diagnostic biomarkers to predict drug resistance in temporal lobe epilepsy: a pilot study. Int J Mol Sci. 2021; 22: 702. https://doi.org/10.3390/ijms22020702.
127. Cui L., Tao H., Wang Y., et al. A functional polymorphism of the microRNA-146a gene is associated with susceptibility to drugresistant epilepsy and seizures frequency. Seizure. 2015; 27: 60–5. https://doi.org/10.1016/j.seizure.2015.02.032.
128. Siddika T., Heinemann I.U. Bringing microRNAs to light: methods for microrna quantification and visualization in live cells. Front Bioeng Biotechnol. 2021; 8: 619583. https://doi.org/10.3389/fbioe.2020.619583.
129. Ye J., Xu M., Tian X., et al. Research advances in the detection of miRNA. J Pharm Anal. 2019; 9 (4): 217–26. https://doi.org/10.1016/j.jpha.2019.05.004.
Шнайдер Наталья Алексеевна, д.м.н., проф.
WoS ResearcherID: M-7084-2014. Scopus Author ID: 24503222300
ул. Бехтерева, д. 3, Санкт-Петербург 192019;
ул. Партизана Железняка, д. 1, Красноярск 660022
Пекарец Николай Александрович
ул. Бехтерева, д. 3, Санкт-Петербург 192019
Пекарец Наталья Игоревна
ул. Красного Восстания, д. 1, Иркутск 664003
Быков Юрий Николаевич, д.м.н., проф.
WoS ResearcherID: S-6938-2016. Scopus Author ID: 57200671414
ул. Красного Восстания, д. 1, Иркутск 664003
Гречкина Виолетта Владимировна
Scopus Author ID: 58076147700
ул. Бехтерева, д. 3, Санкт-Петербург 192019
Дмитренко Диана Викторовна, д.м.н.
WoS ResearcherID: H-7787-2016
ул. Партизана Железняка, д. 1, Красноярск 660022
Петрова Марина Михайловна, д.м.н., проф.
WoS ResearcherID: L-5623-2014. Scopus Author ID: 23987271200
ул. Партизана Железняка, д. 1, Красноярск 660022
Насырова Регина Фаритовна, д.м.н.
WoS ResearcherID: H-7787-2016
ул. Бехтерева, д. 3, Санкт-Петербург 192019;
пр-т Ленина, д. 92, Тула 300012
Шнайдер Н.А., Пекарец Н.А., Пекарец Н.И., Быков Ю.Н., Гречкина В.В., Дмитренко Д.В., Петрова М.М., Насырова Р.Ф. Роль микроРНК как регуляторов системного воспалительного ответа при метаболическом синдроме, индуцированном противоэпилептическими препаратами. Эпилепсия и пароксизмальные состояния. 2025;17(2):208-226. https://doi.org/10.17749/2077-8333/epi.par.con.2025.239
Shnayder N.A., Pekarets N.A., Pekarets N.I., Bykov Yu.N., Grechkina V.V., Dmitrenko D.V., Petrova M.M., Nasyrova R.F. The role of microRNAs as regulators of systemic inflammatory response in anticonvulsant-induced metabolic syndrome. Epilepsy and paroxysmal conditions. 2025;17(2):208-226. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2025.239

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































