


Содержание
Перейти к:
 З. Г. Тадтаева,
А. Н. Галустян,
М. Ю. Кривдина,
В. В. Русановский,
Е. А. Ефет,
А. Е. Кривошеин,
Н. А. Курицына,
О. А. Громова
З. Г. Тадтаева,
А. Н. Галустян,
М. Ю. Кривдина,
В. В. Русановский,
Е. А. Ефет,
А. Е. Кривошеин,
Н. А. Курицына,
О. А. Громова https://doi.org/10.17749/2077-8333/epi.par.con.2022.111
Перейти к:
Миоклоническая эпилепсия с разорванными красными волокнами (англ. myoclonic epilepsy with ragged red fibers, MERRF) – наследуемое по материнской линии заболевание, характеризующееся миоклонической эпилепсией, мозжечковой атаксией и прогрессирующей мышечной слабостью. Его развитие связано с наиболее часто (90% случаев) встречаемой точечной мутацией в позиции 8344 в гене митохондриальной лизиновой транспортной РНК – MTTLys. Диагностика вызывает определенные трудности в связи с недостаточной информированностью практикующих врачей о данной патологии и полиморфизмом клинических проявлений. Проведен краткий обзор литературных данных по современным представлениям о патогенезе заболевания, методам диагностики и возможностям медикаментозного лечения, а также описано клиническое наблюдение синдрома MERRF у ребенка 6 лет, обусловленного точечной мутацией в позиции 8344 в гене MTTLys. Девочка находилась под динамическим наблюдением в психоневрологическом отделении. Выполнено комплексное клинико-лабораторное и инструментальное обследование, включая молекулярно-генетическое тестирование. Представляют интерес прогредиентность и многосимптомность заболевания, легкое повышение лактат-ацидоза при отсутствии типичных нейровизуализационных и электромиографических изменений, что подтверждает необязательность их наличия при MERRF. Отмечено внутрисемейное клиническое разнообразие при отсутствии признаков болезни у матери. Высокоинформативным методом диагностики MERRF является молекулярно-генетическое тестирование. Установление генетического диагноза диктует необходимость медико-генетического консультирования для планирования семьи и предупреждения повторного рождения больного ребенка с наследственной патологией.
Тадтаева З.Г., Галустян А.Н., Кривдина М.Ю., Русановский В.В., Ефет Е.А., Кривошеин А.Е., Курицына Н.А., Громова О.А. Миоклоническая эпилепсия с разорванными красными волокнами в детском возрасте. Эпилепсия и пароксизмальные состояния. 2022;14(1):28-36. https://doi.org/10.17749/2077-8333/epi.par.con.2022.111
Tadtaeva Z.G., Galustyan A.N., Krivdina М.Yu., Rusanovsky V.V., Efet E.A., Krivoshein A.Е., Kuritsyna N.A., Gromova О.A. Myoclonic epilepsy with ragged red fibers in childhood. Epilepsy and paroxysmal conditions. 2022;14(1):28-36. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2022.111
Миоклоническая эпилепсия с разорванными красными волокнами в мышцах (англ. myoclonic epilepsy with ragged red fibers, MERRF) – редкое заболевание из группы митохондриальных энцефаломиопатий, характеризуемое многосистемностью поражения и гетерогенностью фенотипических проявлений (ОМIМ 545000, синонимы: MERRF-синдром, синдром Фукухара, энцефаломиелопатия с разорванными красными волокнами) [1][2]. Заболевание обусловлено структурными нарушениями в ДНК митохондрий.
Термин «разорванные красные волокна» предложен В. Олсоном с соавторами в 1972 г. У пациентов с офтальмоплегией при биопсии мышечной ткани были обнаружены участки скопления пораженных митохондрий на периферии мышечного волокна, которые при окрашивании по методу Гомори приобретали красный цвет. Было сделано предположение, что «разорванные красные волокна» могут быть признаком митохондриальной миопатии.
Первое подробное клиническое описание MERRF принадлежит японскому неврологу Н. Fukuhara (1980 г.), который наблюдал два клинических случая сочетания миоклонической эпилепсии, ассоциированной с «разорванными красными волокнами», с атаксией, снижением интеллекта и лактат-ацидозом в сыворотке крови у пациентов детского и подросткового возраста [3]. Это состояние также известно как синдром Фукухары.
Развитие синдрома MERRF обусловлено точечными мутациями митохондриальной ДНК (мтДНК) в виде замены нуклеотидной последовательности. В настоящее время известно не менее 18 мутаций, ответственных за развитие заболевания [4]. Наиболее частой мутацией (m) в 80–90% случаев является замена аденина на гуанин в положении 8344 в гене митохондриальной транспортной РНК (тРНК) лизина (m. 8344T>C) – ген MTTLys [5]. Эта мутация оказывает воздействие на трансляцию белков, кодируемых мтДНК, нарушает сборку комплексов электронно-транспортной цепи, что приводит к снижению дыхательной функции митохондрий и окислительного фосфорилирования в клетках организма. Учитывая системный дефект энергетического метаболизма, поражаются наиболее энергозависимые органы и ткани с развитием мультисистемности и полиморфизма клинических симптомов. Более редкими являются мутации в том же гене (m. 8356T>C, m.8363G>A, m.8361G>A) [6]. Менее 5% мутаций приходится на гены, кодирующие тРНК, переносящие лейцин, фенилаланин и пролин соответственно, что также приводит к возникновению заболевания [6][7].
Особенностью MERRF является материнский тип наследования. Популяционная частота заболевания в России неизвестна. По данным отдельных эпидемиологических исследований, распространенность MERRF, обусловленного мутацией A8344G, составляет от 0,39 до 1,5 случая на 100 тыс. человек во взрослой популяции ряда европейских стран и 0,25 случая на 100 тыс. человек – среди детского населения на западе Швеции [8–10].
Возраст дебюта варьирует от 3 до 65 лет. Заболевание имеет прогрессирующее течение. Отсутствие патологических проявлений в начале жизни пациента и клинический полиморфизм патологии можно объяснить феноменом гетероплазмии – одновременного существования мутантных и нормальных мтДНК в одной клетке. При клеточном делении распределение мтДНК между дочерними клетками происходит в случайном порядке. Количество мтДНК с возрастом может увеличиваться и постепенно достигать уровня, способного вызвать клиническое проявление заболевания. Неравномерное распределение нормальной и мутированной мтДНК в разных органах и тканях определяет различие симптомов у пострадавших членов семьи.
Наиболее частыми симптомами MERRF являются мозжечковая атаксия, миоклонус, генерализованная эпилепсия, тремор, мышечная слабость. Отличительной особенностью заболевания выступает миоклонус – внезапные подергивания мышечных пучков или их групп в конечностях, лице или туловище, индуцируемые движением или намеренными движениями [1–3]. Миоклонии могут сочетаться с генерализованной эпилепсией, обычно характеризуемой резистентностью к медикаментозному лечению. Развитие эпилептических приступов связывают со снижением продукции энергии аденозинтрифосфата (АТФ), приводящим к нестабильности мембранного потенциала нервной клетки.
Миоклоническая эпилепсия при MERRF входит в группу прогрессирующих миоклонических эпилепсий, к которым также относят болезнь Унферрихта–Лундборга, болезнь Лафора, различные формы нейрональных цероидных липофусцинозов и сиалидозов 1-го и 2-го типов. Наиболее частым типом приступов при MERRF является генерализованный миоклонический припадок. Другие типы приступов, зарегистрированные при MERRF, включают фокальные миоклонические, фокальные клонические и фокальные атонические, генерализованные миоклонические, тонико-клонические, атонические и миоклоническо-атонические припадки или типичные абсансы. У 69% больных миоклоническая эпилепсия сочетается с генерализованными тоникоклоническими приступами. В ряде случаев может развиться эпилептический статус [11].
Мозжечковая атаксия отмечается в более чем 80% наблюдений и может подтверждать дегенеративные изменения спиноцеребеллярного тракта на ранней стадии болезни [12][13]. Для синдрома MERRF характерны также полинейропатия, нейросенсорная тугоухость, кардиомиопатия и/или нарушения сердечного ритма, атрофия зрительного нерва, нарушения со стороны эндокринной и желудочно-кишечной систем, липоматоз, задержка физического развития, непереносимость физических нагрузок, постепенное снижение интеллекта [5]. В настоящее время для оптимизации диагностики и лечения предложен общий алгоритм оценки диагностики митохондриальных заболеваний, который применим также к MERRF [14].
При магнитно-резонансной томографии (МРТ) и компьютерной томографии (КТ) изменения головного мозга малоспецифичны. Обнаруживаются задержка миелинизации, атрофия головного мозга и мозжечка. На Т2-взвешенной томограмме могут выявляться очаги высокой интенсивности белого вещества в области бледного шара, которые могут сочетаться с кальцификацией базальных ганглиев и зубчатого ядра мозжечка [15]. При этом в начале болезни патологические изменения на МРТ могут отсутствовать.
Важным дополняющим и уточняющим нейровизуализационным методом исследования головного мозга является магнитно-резонансная протонная спектроскопия, позволяющая измерить внутриклеточные концентрации таких структурных метаболитов, как холин, N-ацетиласпартат, глютамат/глутамин, лактат, креатин [16]. В исследованиях у пациентов выявляется увеличение уровня молочной кислоты и соотношения холина/креатина в базальных ганглиях на начальных стадиях развития заболевания [17].
Электроэнцефалографическая (ЭЭГ) картина при синдроме MERRF характеризуется эпилептиформной активностью, наличием генерализованных комплексов «спайк–волна», диффузных медленных волн. Реже регистрируется фокальная активность, характерна фотосенситивность [18]. При проведении электронейромиографии отмечается снижение амплитуды и длительности потенциалов двигательных единиц, свидетельствующее о миопатических изменениях [19].
Одним из основных диагностических тестов MERRF является гистологическое исследование мышц. В мышечном биоптате выявляют атрофические изменения. При окрашивании гистологических срезов по методу Гомори наблюдаются «рваные красные волокна» в более чем в 5% мышечных волокон, образующие агломераты по периферии мышечного волокна с содержанием большого количества поврежденных митохондрий, изменением их формы и размеров. Важным гистохимическим маркером является дефицит цитохром-Соксидазы в мышечных волокнах [16].
При биохимическом исследовании крови возможно небольшое повышение уровня молочной и пировиноградной кислоты в крови и/или спинномозговой жидкости даже у бессимптомных членов семьи пациентов [19][20]. В 1,6–8% случаев отмечено повышение белка в ликворе [21]. При первичном обследовании больного выявляют также повышение уровня креатинкиназы в сыворотке крови, которое не является специфичным показателем для митохондриальной миопатии при MERRF, но может свидетельствовать о вовлечении в патологический процесс мышечной системы [22].
Терапия MERRF носит симптоматический характер и направлена на снижение прогрессирования заболевания и улучшение качества жизни пациента. С этой целью больным назначают комбинации препаратов, способствующих улучшению функции митохондрий: коэнзим Q10, идебенон (синтетический аналог СоQ100), левокарнитин, креатина моногидрат, витамины группы В, включая фолиевую кислоту, антиоксиданты (витамины Е и С) [23]. Особое внимание уделяется диете с ограничением количества углеводов.
Пациенты с судорожным синдромом нуждаются в применении противоэпилептических препаратов (ПЭП). Несмотря на внедрение новых ПЭП, лечение эпилепсии при MERRF остается сложной задачей. В настоящее время препаратами выбора для терапии эпилепсии являются леветирацетам, топирамат, зонисамид, пирацетам и бензодиазепины. Противопоказано применение препаратов, влияющих на дыхательную цепь митохондрий [22][24][25]. В частности, вальпроевая кислота вызывает дисфункцию I и IV комплексов дыхательной цепи митохондрий, нарушение образования АТФ и подавление функции ключевых ферментов ß-окисления, приводящее к развитию вторичного карнитинового дефицита. Фенобарбитал ингибирует функцию I комплекса дыхательной цепи, снижает синтез АТФ и мембранный потенциал митохондрий. Разрабатываются также альтернативные методы лечения, включающие кетогенную диету и стимуляцию блуждающего нерва [26].
Клинические случаи MERRF в детском возрасте в доступной отечественной литературе нам не встречались. Представлено собственное клиническое наблюдение MERRF, обусловленного точковой мутацией митохондриальной ДНК m.8344А>G в гене лизиновой транспортной РНК, которое служит иллюстрацией особенностей клинических проявлений и результатов лабораторно-инструментальных методов исследования.
Больная Н., 7 лет, поступила в неврологическое отделение с жалобами на пароксизмы «подкашивания» ног с последующим падением и утратой сознания, «передергивания» в мышцах туловища, изменение походки, тремор верхних конечностей, быструю утомляемость при ходьбе. Девочка находилась под динамическим наблюдением в психоневрологическом отделении. Проводилось комплексное клинико-лабораторное и инструментальное обследование. Молекулярно-генетическое тестирование выполнено в лаборатории наследственных болезней обмена веществ Медико-генетического научного центра (Москва). Разрешение родителей на использование медицинских записей для публикации получено.
Наследственность отягощена по материнской линии: у матери в анамнезе в раннем детстве фебрильный судорожный синдром, у бабушки синдром паркинсонизма (не обследована), двоюродный дядя страдает приступами внезапного падения, нарушена походка (диагноз MERRF при обследовании подтвержден генетически), у троюродного брата прогрессирующая миоклоническая атаксия Ханта и симптоматическая эпилепсия с полиморфными пароксизмами (диагноз MERRF также подтвержден генетически). Больная родилась в срок путем кесарева сечения (многоводие, тазовое предлежание плода). Масса тела при рождении 4100 г, рост 56 см, оценка по шкале Апгар 7/8 баллов. Раннее психомоторное развитие соответствовало возрасту.
Первые признаки заболевания возникли в возрасте 6 лет с появления симптомов нейросенсорной тугоухости (непереносимость громких звуков, снижение слуха, шум в ушах). Через 4 мес появились утомляемость при ходьбе, мозжечковая атаксия, макрография и тремор при письме. Еще через 1 мес присоединились миоклонически-астатические приступы длительностью 2–3 с до 5 раз в сутки.
При видео-ЭЭГ-мониторировании в состоянии сна и бодрствования выявлена эпиактивность с акцентом в теменно-височно-затылочной области справа, при окончательном пробуждении зафиксированы миоклонические приступы.
Назначен леветирацетам в дозе 60 мг/кг/сут. На фоне терапии произошла трансформация приступов в миоклонические до 10 раз в сутки. Для улучшения эффекта в терапию добавлена пролонгированная форма вальпроевой кислоты. Отмечено урежение приступов до 1 раза в сутки, но наросла очаговая неврологическая симптоматика: усилилась атаксия, появились когнитивные нарушения (снижение памяти и скорости мышления), ухудшилась речь (дизартрия).
Учитывая типичный синдромокомплекс, характерный для митохондриальных заболеваний, рекомендовано генетическое обследование. При поступлении в психоневрологическое отделение проведена коррекция терапии с учетом результатов молекулярно-генетического исследования (выявлена точковая мутация митохондриальной ДНК m.8344А>G в гене tRNA-Lys в гомозиготном состоянии). Добавлены топирамат на фоне постепенной отмены пролонгированной формы вальпроевой кислоты, метаболическая и антиоксидантная терапия, что способствовало стабилизации состояния и урежению миоклоний до 1 раза в месяц.
При поступлении состояние средней тяжести. Соматический статус без особенностей. При внешнем осмотре наблюдается отставание в физическом развитии (масса тела 23,5 кг, рост 127 см).
Неврологический статус: нарушение речевых функций, дизартрия средней степени выраженности, дисфония, смешанная дислексия. Сглажена носогубная складка слева. Мышечный тонус верхних конечностей повышен по типу «зубчатого колеса». Сухожильные рефлексы в конечностях повышены, симметричные, D=S. Патологических стопных знаков нет. Двусторонний клонус стоп. При ходьбе атаксия, больная несколько отклоняется вправо. Интенционный тремор и мимопопадание при выполнении пальценосовой пробы. Гиперкинезы: миоклонии мышц верхней части туловища. Нарушения чувствительности не выявлено. Менингеальные симптомы отрицательные. Высшие корковые функции: словарный запас ограничен, со школьной общеобразовательной программой не справляется.
Анализ крови
Клинический анализ крови без патологии. Биохимический анализ крови: лактат 4,7 ммоль/л (норма 0,00– 2,70 ммоль/л), глюкоза 4,40 ммоль/л (норма 3,30–5,50 ммоль/л), увеличение активности сердечного изофермента креатинкиназы МВ 7,7 нг/мл (норма 0,0–3,4 нг/мл)
Магнитно-резонансная томография
При проведении МРТ головного мозга (Philips Ingenia 1.5Т, Нидерланды) выявлена смешанная гидроцефалия заместительного характера. При протонной МР-спектроскопии значимых пиков липида и лактата не выявлено. Метаболические изменения в виде повышения пиков холина в белом веществе на уровне базальных ядер расценили как признаки продолжающегося незавершенного этапа миелинизации.
Электроэнцефалография
Исследование проведено на аппарате «Мицар-ЭЭГ» («Мицар», Россия). Возрастной ритм замедлен, деформирован. Нерегулярное региональное замедление биоэлектрической активности головного мозга в виде усиления тета-ритма в теменно-затылочных отведениях обоих полушарий. Генерализованные билатеральные вспышки эпилептиформной активности, высокоамплитудные комплексы «острая – медленная волна» (рис. 1).
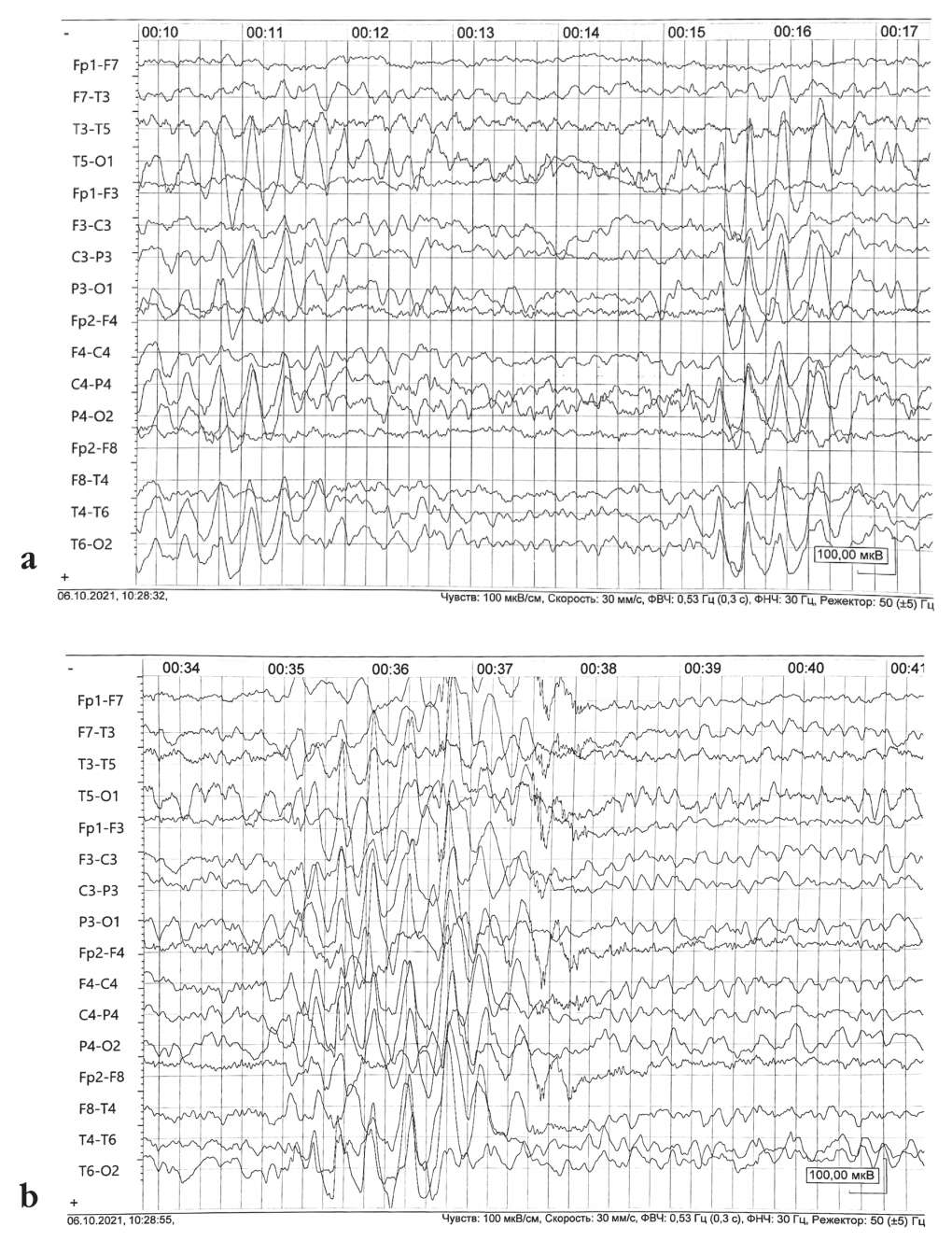
Рисунок 1. Электроэнцефалограммы пациентки Н. (7 лет): а – нерегулярное региональное замедление биоэлектрической активности головного мозга в виде усиления тета-ритма в теменно-затылочных отведениях обоих полушарий; b – генерализованные билатеральные вспышки эпилептиформной активности, высокоамплитудные комплексы «острая – медленная волна»
Figure 1. Electroencephalograms of patient N. (7 years old): а – irregular regional deceleration of bioelectric brain activity in the form of an increase in theta rhythm frequency in parietal-occipital leads of both hemispheres; b – generalized bilateral outbreaks of epileptiform activity, high-amplitude acute-slow wave complexes
Электронейромиография
При проведении электронейромиографии признаков поражения сенсорных и моторных волокон нервов верхних и нижних конечностей не выявлено.
Электрокардиография
Синусовый ритм с частотой сердечных сокращений 101 уд/мин. Ритм умеренно частый. Нормальное положение электрической оси сердца. Метаболические изменения с умеренным нарушением процессов реполяризации по нижнебоковой стенке левого желудочка. Эхокардиография без патологии.
Генетическое исследование
При генетическом исследовании выявлена точковая мутация митохондриальной ДНК m.8344А>G в гене tRNA-Lys в состоянии гомоплазмии. Таким образом, на основании полученного генетического обследования ребенку выставлен диагноз: синдром MERRF, обусловленный мутацией в гене митохондриальной лизиновой тРНК MTTLy.
MERRF – редкое митохондриальное заболевание, наследуемое по материнской линии, которое характеризуется мультисистемностью поражения и полиморфизмом клинических проявлений [1][6]. Основными диагностическими критериями MERRF-синдрома являются: материнский тип наследования, наличие собственного митохондриального генома, кодирующего синтез полипептидов, образующих дыхательную цепь, гетероплазмия, значительный полиморфизм клинических проявлений с прогрессированием симптоматики и различными сроками манифестации, внутрисемейное разнообразие симптомов у ближайших родственников по материнской линии [14].
Заболевание чаще дебютирует в детском и подростковом возрасте, что подтверждается клиническим наблюдением синдрома MERRF, описанным N. Fukuhara [3][8]. Особенностью дебюта в нашем наблюдении было развитие нейросенсорной тугоухости. Чаще первыми симптомами являются миоклонии, атаксия, генерализованная эпилепсия [2]. В представленном клиническом наблюдении заболевание характеризовалось наличием у пациентки гиперкинетического синдрома (миоклоний) с последующим развитием генерализованных эпилептических припадков, мозжечковой атаксии, отставанием в физическом развитии.
Нейровизуализационными признаками поражения головного мозга при синдроме MERRF являются гиперинтенсивные очаги в Т2-режиме, не соответствующие зоне кровоснабжения мозговых артерий в подкорковых областях головного мозга, которые могут сочетаться с кальцификацией базальных ганглиев [12][15]. Универсальными диагностическими тестами, подтверждающими нарушение функций митохондрий, являются повышение уровней лактата и пирувата в сыворотке крови и ликворе, а также гистохимическое исследование биоптатов мышц пациентов, которое выявляет характерные для данного заболевания изменения мышечных волокон – «рваные красные волокна», содержащие комплексы поврежденных митохондрий по периферии мышечного волокна [14]. В нашем наблюдении отмечено лишь легкое повышение лактатацидоза при отсутствии специфических изменений на электронейромиограмме и при нейровизуализации [2].
Наличие полиморфной симптоматики с прогредиентным течением позволило заподозрить митохондриальное заболевание и обосновать молекулярногенетическое исследование, которое в настоящее время признано основным методом диагностики митохондриальных энцефаломиопатий [14]. В нашем случае у пациентки выявлена типичная точковая мутация митохондриальной ДНК m.8344А>G, характерная для MERRF. Генетическая диагностика заболевания способствовала назначению адекватной терапии с применением препаратов, повышающих активность дыхательной цепи, – кофакторов ферментных реакций энергетического обмена, антиоксидантов и средств, улучшающих метаболизм мышечной ткани [23].
Применение ПЭП при эпилепсии имеет особенности, связанные с митохондриальной «токсичностью», поэтому их не следует использовать при MERRF. Наиболее выраженное токсическое действие на митохондриальную функцию оказывают вальпроевая кислота, карбамазепин, фенитоин и фенобарбитал [22][24][26]. Назначенная комбинированная терапия привела к улучшению состояния пациентки и значительному урежению эпилептических припадков.
Внедрение молекулярно-генетических методов исследования позволяет установить нозологическую форму митохондриальных энцефаломиопатий и способствует пониманию патогенеза MERRF и ее адекватной терапии. Установление генетического диагноза также диктует необходимость медико-генетического консультирования для планирования семьи и предупреждения повторного рождения больного ребенка с наследственной патологией.
1. Айкарди Ж., Бакс М., Гиллберг К. (ред.) Заболевания нервной системы у детей. М.: БИНОМ; 2013: 259–360.
2. Руденская Г.Е., Захарова Е.Ю. Наследственные нейрометаболические болезни юношеского и взрослого возраста. M.: ГЭОТАР-Медиа; 2018: 377.
3. Fukuhara N., Tokiguchi S., Shirakawa K., Tsubaki T. Myoclonus epilepsy associated with ragged-red fibers (mitochondrial abnormalities): disease entity or a syndrome. J Neurol Sci. 1980; 47 (1): 117–33. http://doi.org/10.1016/0022-510x(80)90031-3.
4. Lorenzoni P.J., Scola R.H., Kay C.S., et al. When should MERRF (myoclonus epilepsy associated with with ragged red fibers) be the diagnosis? Arq Neuropsiquiatr. 2014; 72 (10): 803–11. http://doi.org/10.1590/0004-282x20140124.
5. Mancuso M., Orsucci D., Angelini C., et al. Phenotypic heterogeneity of the 8344A > G mtDNA “MERRF” mutation. Neurology. 2013; 80 (22): 2049–54. http://doi.org/10.1212/WNL.0b013e318294b44c.
6. Finsterer J., Kovacs G.G. Psoriasis, hyperlipidemia, bulbar involvement, and diarrhoea in MERRF-syndrome due to the m. 8344A> G tRNA (Lys) mutation. Iran J Neurol. 2017; 16 (1): 45–9.
7. Liu K., Zhao H., Ji K., et al. MERRF/MELAS overlap syndrome due to the m.3291T>C mutation. Metab Brain Dis. 2014; 29 (1): 139–44. https://doi.org/10.1007/s11011-013-9464-5.
8. Darin N., Oldfors A., Moslemi A.R., et al. The incidence of mitochondrial encephalomyopathies in childhood: clinical features and morphological, biochemical, and DNA abnormalities. Ann Neurol. 2001; 49 (3): 377–83. https://doi.org/10.1002/ana.75.
9. Schaefer A.M., McFarland R., Blakely E.L., et al. Prevalence of mitochondrial DNA disease in adults. Ann Neurol. 2008; 63 (1): 35–9. https://doi.org/10.1002/ana.21217.
10. Remes A.M., Majamaa-Voltti K., Kärppä M., et al. Prevalence of large-scale mitochondrial DNA deletions in an adult Finnish population. Neurology. 2005; 64 (6): 976–81. https://doi.org/10.1212/01.WNL.0000154518.31302.ED.
11. Новиков А.Е. Эволюция в клинической эпилептологии. М.: Флинта; 2015: 583 с.
12. Ito S., Shirai W., Asahina M., et al. Clinical and brain MR imaging features focusing on the brain stem and cerebellum in patients with myoclonic epilepsy with ragged-red fibers due to mitochondrial A8344G mutation. AJNR Am J Neuroradiol. 2008; 29 (2): 392–5. https://doi.org/10.3174/ajnr.A0865.
13. Finsterer J. Mitochondrial ataxias. Can J Neurol Sci. 2009; 36 (5): 543–53. http://doi.org/10.1017/s0317167100008027.
14. Иллариошкин С.Н. Алгоритм диагностики митохондриальных энцефаломиопатий. Атмосфера. Нервные болезни. 2007; 3: 23–7.
15. Ублинский М.В., Манжурцев А.В., Меньщиков П.Е. и др. Мультимодальные исследования головного мозга человека с использованием функциональной магнитно-резонансной томографии и магнитно-резонансной спектроскопии. Анналы клинической и экспериментальной неврологии. 2018; 12 (1): 54–60.
16. Betts J., Lightowlers R.N., Turnbull D.M. Neuropathological aspects of mitochondrial DNA disease. Neurochem Res. 2004; 29 (3): 505–11. https://doi.org/10.1023/b:nere.0000014821.07269.8d.
17. Chuang C.S., Lo M.C., Lee K.W., Liu C.S. Magnetic resonance spectroscopy study in basal ganglia of patients with myoclonic epilepsy with ragged-red fibers. Neurol India. 2007; 55 (4): 385–7. https://doi.org/10.4103/0028-3886.37096.
18. Sinha S., Satishchandra P., Gayathri N., et al. Progressive mioclonic epilepsy: a clinical, electrophysiological and pathological study from South India. J Neurol Sci. 2007; 252 (1): 16–23. http://doi.org/10.1016/j.jns.2006.09.021.
19. DiMauro S., Hirano M., Kaufmann P., et al. Clinical features and genetics of myoclonic epilepsy with ragged red fibers. Adv Neurol. 2002; 89: 217–29.
20. Zeviani M., Amati P., Bresolin N., et al. Rapid detection of the A-to-G(8344) mutation of mtDNA in Italian families with myoclonus epilepsy and ragged-red fibers (MERRF). Am J Hum Genet. 1991; 48 (2): 203–11.
21. Marotta R., Chin J., Quigley A., et al. Diagnostic screening of mitochondrial DNA mutations in Australian adults 1990–2001. Intern Med J. 2004; 34 (1-2): 10–9. https://doi.org/10.1111/j.1444-0903.2004.t01-3-.x.
22. Finsterer J., Zarrouk-Mahjoub S. Management of epilepsy in MERRF syndrome. Seizure. 2017; 50: 166–70. http://doi.org/10.1016/j.seizure.2017.06.010.
23. Николаева Е.А., Яблонская М.И., Харабадзе М.Н. и др. Эффективность комплексной терапии при разных формах митохондриальных заболеваний у детей. Российский вестник перинатологии и педиатрии. 2009; 54 (6): 26–30.
24. Заваденко Н.Н., Холин А.А. Эпилепсия у детей с митохондриальными заболеваниями: особенности диагностики и лечения. Эпилепсия и пароксизмальные состояния. 2012; 4 (2): 21–7.
25. Lorenzoni P.J., Scola R.H., Kay C.S., et al. MERRF: clinical features, muscle biopsy and molecular genetics in Brazilian patients. Mitochondrion. 2011; 11 (3): 528–32. http://doi.org/10.1016/j.mito.2011.01.003.
26. Finsterer J., Zarrouk Mahjoub S. Mitochondrial toxicity of antiepileptic drugs and their tolerability in mitochondrial disorders. Expert Opin Drug Metab Toxicol. 2012; 8 (1): 71–9. https://doi.org/10.1517/17425255.2012.644535.
Тадтаева Зара Григорьевна – доктор медицинских наук, профессор кафедры фармакологии с курсом клинической фармакологии и фармакоэкономики.
ул. Литовская, д. 2, Санкт-Петербург 194100.
РИНЦ SPIN-код: 6086-0169
Галустян Анна Николаевна – кандидат медицинских наук, доцент, заведующая кафедрой фармакологии с курсом клинической фармакологии и фармакоэкономики.
ул. Литовская, д. 2, Санкт-Петербург 194100.
РИНЦ SPIN-код: 3303-7650
Кривдина Марина Юрьевна – невролог психоневрологического отделения.
ул. Литовская, д. 2, Санкт-Петербург 194100.
Русановский Владимир Васильевич – доктор медицинских наук, профессор кафедры фармакологии с курсом клинической фармакологии и фармакоэкономики.
ул. Литовская, д. 2, Санкт-Петербург 194100.
РИНЦ SPIN-код: 7010-4530
Ефет Елена Анатольевна – кандидат медицинских наук, заведующая психоневрологическим отделением.
ул. Литовская, д. 2, Санкт-Петербург 194100.
Кривошеин Александр Евгеньевич – студент.
ул. Литовская, д. 2, Санкт-Петербург 194100.
Курицына Наталия Андреевна – кандидат медицинских наук, доцент кафедры фармакологии с курсом клинической фармакологии и фармако-экономики.
ул. Литовская, д. 2, Санкт-Петербург 194100.
РИНЦ SPIN-код: 4361-7365
Громова Ольга Алексеевна – доктор медицинских наук, профессор, научный руководитель.
ул. Вавилова, д. 4, Москва 119333.
Scopus Author ID: 7003589812
WoS ResearcherID: J-4946-2017
РИНЦ SPIN-код: 6317-9833
Тадтаева З.Г., Галустян А.Н., Кривдина М.Ю., Русановский В.В., Ефет Е.А., Кривошеин А.Е., Курицына Н.А., Громова О.А. Миоклоническая эпилепсия с разорванными красными волокнами в детском возрасте. Эпилепсия и пароксизмальные состояния. 2022;14(1):28-36. https://doi.org/10.17749/2077-8333/epi.par.con.2022.111
Tadtaeva Z.G., Galustyan A.N., Krivdina М.Yu., Rusanovsky V.V., Efet E.A., Krivoshein A.Е., Kuritsyna N.A., Gromova О.A. Myoclonic epilepsy with ragged red fibers in childhood. Epilepsy and paroxysmal conditions. 2022;14(1):28-36. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2022.111

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































