



Содержание
Перейти к:
https://doi.org/10.17749/2077-8333/epi.par.con.2022.123
Перейти к:
До недавнего времени не было официально утвержденной Международной Противоэпилептической Лигой (англ. International League Against Epilepsy, ILAE) классификации эпилептических синдромов. В 2022 г. в результате многолетних упорных усилий экспертов и общественности вышли в свет работы, представляющие определение и классификацию эпилептических синдромов. ILAE утвердила следующее определение эпилептического синдрома: «характерный набор клинических и электроэнцефалографических признаков, часто обусловленных специфическими этиологическими факторами (структурными, генетическими, метаболическими, иммунными и инфекционными)». Классификация эпилептических синдромов выполнена по возрастному принципу: с началом в неонатальном периоде и младенчестве, с началом в детском возрасте, с началом в разном возрасте. Отдельно были выделены синдромы идиопатической генерализованной эпилепсии. Клинические данные по каждому эпилептическому синдрому приведены в едином шаблоне: эпидемиология, клиническая картина, анамнез заболевания, тип(ы) приступов, данные электроэнцефалографии, результаты нейровизуализации, данные генетических исследований, результаты других лабораторных исследований (когда они информативны), дифференциальный диагноз. Приводятся критерии постановки диагноза, включающие обязательные критерии, настораживающие признаки и критерии исключения. Эта классификация должна стать отправной точкой для дальнейшего улучшения организации работы практикующих специалистов, которые занимаются проблемой эпилепсии.
Блинов Д.В. Эпилептические синдромы: определение и классификация Международной Противоэпилептической Лиги 2022 года. Эпилепсия и пароксизмальные состояния. 2022;14(2):101-182. https://doi.org/10.17749/2077-8333/epi.par.con.2022.123
Blinov D.V. Epilepsy syndromes: the 2022 ILAE definition and classification. Epilepsy and paroxysmal conditions. 2022;14(2):101-182. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2022.123
Эпилепсия является одним из наиболее распространенных неврологических заболеваний, встречающимся на разных этапах жизни у мужчин и женщин, но чаще всего (а именно более чем в 60 случаях на 100 тыс. населения) – у детей в возрасте младше 5 лет, а также в старшем (от 65 лет) возрасте [1][2].
При одном из наиболее высоких количестве неврологов (19,4 на 100 тыс. населения) в России остаются нерешенными вопросы междисциплинарного взаимодействия и маршрутизации пациентов с эпилепсией [3–6]. Проблемы с доступом к диагностике и лечению признаются и во всем мире: 116 государств – членов Всемирной организации здравоохранения единогласно одобрили Межсекторальный глобальный план действий по борьбе с эпилепсией и другими неврологическими расстройствами (англ. Intersectoral Global Action Plan on Epilepsy and Other Neurological Disorders, IGAP), согласно которому к 2031 г. необходимо увеличить охват мерами по лечению эпилепсии на 50% по сравнению с таковым в 2021 г. Для этого 80% стран должны обновить законодательство с целью защиты прав пациентов с эпилепсией и улучшения доступа к службам здравоохранения с тем, чтобы 90% страдающих эпилепсией знали о своем диагнозе как о поддающемся лечению заболевании, 80% из них были доступны соответствующие безопасные антиэпилептические препараты (АЭП), а 70% получали лечение, позволяющее достичь адекватного контроля над приступами [7][8].
Одним из основных разработчиков IGAP являлась наиболее авторитетная организация, занимающаяся проблемой эпилепсии по всему миру, – Международная Противоэпилептическая Лига (англ. International League Against Epilepsy, ILAE), имеющая свыше 100 тыс. членов и национальные отделения более чем в 100 странах. В 2017 г. ILAE обновила определение эпилепсии, представила рабочую классификацию типов приступов и новую классификацию эпилепсии [9–11]. Классификация ILAE 2017 г. содержит три диагностических уровня – тип приступа, тип эпилепсии и эпилептический синдром, при этом на каждом уровне необходимо учитывать этиологию и сопутствующие (коморбидные) состояния. Определение эпилептического синдрома является важным для уточнения этиологии, методов лечения и прогноза, однако тогда классификация синдромов эпилепсии разработана и официально утверждена не была.
В 2017 г. в рамках ILAE была создана Рабочая группа по классификации и определениям (англ. Nosology and Definitions Task Force), которая продолжила работу по формированию дефиниции эпилептического синдрома, описанию и классификации эпилептических синдромов с использованием терминологии классификации типов приступов и эпилепсии 2017 г. [9–13]. В мае 2022 г. новая Классификация эпилептических синдромов ILAE как результат усилий Рабочей группы по классификации и определениям вышла в свет.
Группирование эпилептических синдромов было выполнено по принципу возраста возникновения – начала или дебюта: с началом в неонатальном периоде и младенчестве (младше 2 лет), с началом в детском возрасте (2–12 лет), с началом в разном возрасте (т.е. дебют может произойти как у детей (18 лет и младше), так и у взрослых (19 лет и старше)). Отдельно были выделены синдромы идиопатической генерализованной эпилепсии. В соответствии с Классификацией эпилепсии ILAE 2017 г. синдромы в каждой из возрастных групп были дополнительно разделены на генерализованные, фокальные или генерализованные и фокальные в зависимости от типа приступов (т.е. эпилептические синдромы, имеющие черты как фокальных, так и генерализованных). Также была выделена отдельная категория для эпилептических синдромов с эволюционной и/или эпилептической энцефалопатией, т.е. когда она связана либо с основной этиологией, либо с наложенной эпилептической активностью, либо с тем и другим (англ. developmental and/or epileptic encephalopathy, DEE), и синдромов с прогрессирующим ухудшением неврологического статуса.
Одним из принципов построения новой классификации является использование описательных названий синдромов, однако были сохранены названия «синдром Драве» и «синдром Леннокса–Гасто». Причины состоят в том, что они могут включать в себя несколько типов приступов (а синдром Леннокса–Гасто включает несколько этиологий, которые было бы сложно описать кратким названием), а также в том, что эти названия повсеместно используются в клинических исследованиях, при регистрации и назначении орфанных препаратов [13][14]. Кроме того, был сохранен термин «синдром Расмуссена», поскольку Рабочая группа ILAE не смогла предложить альтернативу этому хорошо зарекомендовавшему себя названию, которое охватывает эпилепсию, различные особенности визуализации и прогрессирующее ухудшение неврологического статуса, наблюдаемое при указанном состоянии [15].
Клинические данные по каждому эпилептическому синдрому сводились в единый шаблон, который содержал:
В таком шаблоне члены Рабочей группы ILAE подготовили проекты описания каждого из эпилептических синдромов, после чего они обсуждались на онлайн- и очных встречах, в т.ч. совместно с Американским обществом эпилепсии (англ. American Epilepsy Society) в 2018 и 2019 гг., на Европейском эпилептологическом конгрессе (англ. European Congress of Epileptology) в 2018 г. и на Международном конгрессе по эпилепсии (англ. International Epilepsy Congress) в 2019 г. Далее проекты дорабатывались членами Рабочей группы ILAE с учетом полученных комментариев [13].
Для достижения консенсуса использовали модифицированный дельфийский метод экспертной оценки [16]. Для каждого синдрома согласно описанному выше шаблону были предложены основные критерии, разделенные на следующие группы:
Для отдельных синдромов к оценке были предложены и дополнительные определения: возможность развития синдрома, или синдром в процессе развития (термин для обозначения синдромов, при которых в начале отсутствуют все обязательные диагностические признаки, но требуется время для их развития, например ранняя стадия синдрома Расмуссена), и синдром без лабораторного подтверждения (если отсутствует доступ к ЭЭГ, магнитно-резонансной томографии (МРТ) или другим исследованиям, без которых может оказаться невозможным с достаточной уверенностью диагностировать некоторые синдромы) [12][13].
Каждый участник обсуждения заполнял онлайн-опросник, в котором оценил все предложенные критерии по 9-балльной шкале Лайкерта (1 балл – «абсолютно не согласен», 9 баллов – «абсолютно согласен»). По любому критерию, оцененному ниже 7 баллов, участникам предлагалось оставить комментарии в произвольной форме. В дальнейшую работу включались только критерии, набравшие в среднем 7 баллов и более, в то время как остальные дополнительно рассматривались Рабочей группой ILAE с тщательным учетом поступивших комментариев. Всего состоялось несколько раундов обсуждения с использованием дельфийского метода.
Подготовленные статьи с изложением позиции ILAE по определениям и классификации эпилептических синдромов были предложены для общественного обсуждения на официальном сайте ILAE, а также рецензентам журнала Epilepsia с использованием раундов дельфийского метода, после чего они были финализированы членами Рабочей группы ILAE [12][13][16].
ILAE утвердила следующее определение: «Эпилептический синдром – характерный набор клинических и ЭЭГ-признаков, часто обусловленных специфическими этиологическими факторами (структурными, генетическими, метаболическими, иммунными и инфекционными)» [12][13].
В своей основе эпилептический синдром должен иметь характерные электроклинические признаки (рис. 1). Постановка синдромального диагноза у человека с эпилепсией часто связана с изменением прогноза и подходов к ведению пациента. Эпилептические синдромы, как правило, имеют возрастные особенности и ряд специфических сопутствующих заболеваний [13]. Они обычно начинаются в определенном возрасте, а в некоторых случаях и самокупируются в определенном возрасте. Для многих эпилептических синдромов установлена корреляция с рядом специфических интеллектуальных, психических и других сопутствующих заболеваний, тогда как для других характерным признаком является их отсутствие.

Рисунок 1. Характеристики эпилептического синдрома (адаптировано из [12])
Figure 1. An epileptic syndrome characteristics (adapted from [12])
В группу новорожденных и младенцев входят как недоношенные, так и доношенные дети от момента рождения до 2-летнего возраста. Недавнее проспективное исследование продемонстрировало, что наиболее высокие показатели заболеваемости эпилепсией отмечаются в группах детей в возрасте до 6 мес (75 случаев на 100 тыс. живорождений) и от 6 до 12 мес (62 случая на 100 тыс. живорождений). Это выше, чем в предыдущих ретроспективных исследованиях [14][17]. При начале эпилепсии в раннем возрасте отмечаются выраженные когнитивные расстройства, сопутствующие заболевания, которые чаще развиваются у детей с большим количеством приступов и фармакорезистентной эпилепсией. Через 2 года после постановки диагноза до 50% из них имеют общую задержку развития [14][17–22]. Синдромальный диагноз эпилепсии у детей младше 3 лет может быть поставлен не менее чем в 54% случаев при использовании новейших методов нейровизуализации и генетических исследований [14][17].
Возникающие в неонатальном периоде и младенчестве эпилептические синдромы разделены на две основные группы (рис. 2):

Рисунок 2. Классификация эпилептических синдромов ILAE 2022 г.: эпилептические синдромы с началом в неонатальном периоде и младенчестве (адаптировано из [14])
Figure 2. The 2022 ILAE Classification of Epilepsy Syndromes: epilepsy syndromes with onset in neonates and infants (adapted from [14])
Также выделена группа этиологически специфичных эпилептических синдромов, большинство из которых относятся к эволюционным и эпилептическим энцефалопатиям. Их развитие обусловлено специфической генетической, структурной, метаболической, иммунной или инфекционной этиологией и имеет последовательные электроклинические особенности, специфическое лечение и прогноз.
В группе самокупирующихся эпилептических синдромов выделены самокупирующаяся (семейная) неонатальная эпилепсия (англ. self-limited (familial) neonatal epilepsy, SeLNE), самокупирующаяся семейная неонатально-младенческая эпилепсия (англ. self-limited familial neonatal-infantile epilepsy, SeLFNIE) и самокупирующаяся (семейная) младенческая эпилепсия (англ. self-limited (familial) infantile epilepsy, SeLIE), имеющие в целом схожие электроклинические признаки, а также генетическая эпилепсия с фебрильными судорогами плюс (англ. genetic epilepsy with febrile seizures plus, GEFS+) и миоклоническая эпилепсия младенчества (англ. myoclonic epilepsy in infancy, MEI).
При самокупирующихся эпилептических синдромах, начинающихся в возрасте младше 2 лет, приступы, как правило, хорошо корректируются АЭП, а когнитивные функции не снижены или имеют место незначительные когнитивные нарушения. В группе самокупирующихся эпилепсий есть синдромы, при которых как de novo, так и наследственные варианты патогенеза вызывают в целом сходные электроклинические признаки и в семейных, и в несемейных случаях. Поэтому авторы классификации особо оговаривают, что это вторичный признак, который может использоваться тогда, когда это уместно, и приводят его не всегда, а также в скобках после основного термина «самокупирующийся» [14][23].
В группе DEE выделены ранняя младенческая эволюционная и эпилептическая энцефалопатия (англ. early infantile developmental and epileptic encephalopathy, EIDEE) с началом исключительно в возрасте до 3 мес и другие синдромы, которые обычно проявляются после 3 мес или имеют дебют как в раннем, так и в позднем младенческом периоде: эпилепсия младенчества с мигрирующими фокальными приступами (англ. epilepsy in infancy with migrating focal seizures, EIMFS), синдром инфантильных эпилептических спазмов (англ. infantile epileptic spasms syndrome, IESS), а также синдром Драве. При DEE сопутствующие заболевания нервной системы могут быть связаны как с основной причиной, так и с негативными последствиями неконтролируемой эпилептической активности [14].
Термин IESS был введен в качестве наиболее полно отражающего характерный тип приступов, что полезно для ранней диагностики и своевременного назначения соответствующей терапии. Во многих случаях не отмечается наличия полной триады синдрома Веста (может отсутствовать гипсаритмия или регресс), поэтому в новой классификации предлагается использовать термин IESS. Поскольку существуют совпадения электроклинических данных при синдроме Отахара и ранней миоклонической энцефалопатии, оба имеют общую генетическую и структурную этиологию, а также во многих случаях наблюдается неполное соответствие критериям, данные два синдрома были объединены в один синдром EIDEE [14].
К группе эпилептических синдромов со специфичной этиологией, или этиологически специфичных синдромов, отнесены устойчивые электроклинические фенотипы, имеющие сильную ассоциацию с конкретной этиологией. На данном этапе не идентифицированы все этиологически специфичные эпилептические синдромы, но представлены определения для их ограниченного числа, включая DEE, связанные с мутациями в генах KCNQ2, CDKL5, PCDH19, SCL2A1, пиридоксини пиридокс(ам)ин-5'фосфат-зависимые эпилепсии, эпилепсии, обусловленные недостаточностью переносчика глюкозы 1-го типа (GLUT1), синдром Штурге–Вебера и геластические приступы с гипоталамической гамартомой [14].
Эпилептический синдром у детей представляется возможным выявить по крайней мере в 1/3 случаев эпилепсии [24–26]. Эпилептические синдромы детского возраста условно делятся на три основные группы [26]:
Распознавание эпилептических синдромов детского возраста требует тщательного анализа семиологии, эволюции приступов во времени, хода развития ребенка, а также данных ЭЭГ, включая фоновые, интериктальные и иктальные паттерны, а в некоторых случаях – МРТ и генетических исследований. Иногда они могут развиваться из других синдромов или типов эпилепсии. Например, синдром инфантильных эпилептических спазмов может развиться в синдром Леннокса–Гасто, а структурная фокальная эпилепсия или самокупирующаяся эпилепсия с центрально-темпоральными спайками (англ. self-limited epilepsy with centrotemporal spikes, SeLECTS), ранее известная как «доброкачественная роландическая эпилепсия» или «доброкачественная эпилепсия с центрально-темпоральными спайками», – в EE-SWAS. У детей с предшествующим нормальным развитием может развиться тяжелая острая энцефалопатия с последующей фармакорезистентной эпилепсией – FIRES или HHE. Более того, SeLFE может частично совпадать с идиопатической генерализованной эпилепсией или даже эволюционировать в нее, отражая лежащую в основе патогенеза предрасположенность пациента к появлению эпилептических приступов [26][27].
На SeLFE приходится до 25% всех эпилептических синдромов детского возраста. Важную роль в этиологии SeLFE играют генетические факторы. SeLFE развивается у здоровых в остальном детей, характеризуется отсутствием патологии в когнитивной сфере и неврологическом статусе. Почти у всех больных к пубертатному периоду наступает ремиссия. Семиология приступов и ЭЭГ-особенности специфичны для каждого из синдромов, входящих в эту группу. Согласно Классификации эпилептических синдромов ILAE 2022 г. исходя из долгосрочного прогноза в рамках SeLFE выделяют две группы синдромов (рис. 3).

Рисунок 3. Классификация эпилептических синдромов ILAE 2022 г.: самокупирующиеся фокальные эпилепсии детского возраста (адаптировано из [26]). ЭЭГ – электроэнцефалография
Figure 3. The 2022 ILAE Classification of Epilepsy Syndromes: self-limited focal epilepsies of childhood (adapted from [26]). EEG – electroencephalography
В первую группу (группа 1) входят два синдрома: самокупирующаяся эпилепсия с вегетативными (автономными) приступами (англ. self-limited epilepsy with autonomic seizures, SeLEAS), ранее именовавшаяся как «синдром Панайотопулоса» или «доброкачественная затылочная эпилепсия с ранним началом», и самокупирующаяся эпилепсия с центрально-темпоральными спайками (англ. self-limited epilepsy with centrotemporal spikes, SeLECTS). Синдромы SeLEAS и SeLECTS имеют следующие общие черты:
Вторая группа (группа 2) включает два синдрома: детская затылочная зрительная эпилепсия (англ. childhood occipital visual epilepsy, COVE), ранее именовавшаяся как «синдром поздней доброкачественной затылочной эпилепсии» или «идиопатическая детская затылочная эпилепсия типа Гасто», и фотосенситивная затылочная эпилепсия (англ. photosensitive occipital lobe epilepsy, POLE), ранее известная как «идиопатическая фотосенситивная затылочная эпилепсия».
На рисунке 3 представлены возраст начала и данные ЭЭГ для каждого из синдромов. В первой группе ремиссия ожидается во всех случаях. Во второй группе высока вероятность ремиссии, однако у некоторых пациентов приступы могут сохраняться после подросткового возраста [26].
К синдромам генетической генерализованной эпилепсии детского возраста согласно Классификации эпилептических синдромов ILAE 2022 г. отнесены (рис. 4): детская абсансная эпилепсия (англ. childhood absence epilepsy, CAE), эпилепсия с миоклонией век (англ. epilepsy with eyelid myoclonia, EEM) и эпилепсия с миоклоническими абсансами (англ. epilepsy with myoclonic absence, ЕМА). Присутствующая на рисунке EMAtS относится к эволюционным и/или эпилептическим энцефалопатиям.

Рисунок 4. Классификация эпилептических синдромов ILAE 2022 г.: синдромы генетической генерализованной эпилепсии детского возраста (адаптировано из [26]). ЭЭГ – электроэнцефалография; DEE (англ. developmental and epileptic encephalopathies) – эволюционная и/или эпилептическая энцефалопатия
Figure 4. The 2022 ILAE Classification of Epilepsy Syndromes: genetic generalized epilepsy syndromes of childhood (adapted from [26]). EEG – electroencephalography; DEE – developmental and/or epileptic encephalopathies
Наиболее часто встречающимся и хорошо изученным из них является CAE, который относится к идиопатической генерализованной эпилепсии. EEM и EMA имеют более серьезный прогноз, чем CAE, и отличаются более высокой долей фармакорезистентности к АЭП и сопутствующих заболеваний когнитивной сферы. В семейном анамнезе часто отмечается наличие идиопатической генетической эпилепсии или GEFS+. При EMAtS обычно наблюдается застой или регресс в развитии, поэтому данный синдром в классификации отнесен к DEE [26].
Согласно Классификации эпилептических приступов ILAE 2022 г. к данным эпилептическим синдромам относятся EMAtS, синдром Леннокса–Гасто, эволюционная и/или эпилептическая энцефалопатия в сочетании со спайк-волновой активностью во сне (англ. developmental and/or epileptic encephalopathy in combination with spikeandwave activation in sleep, DEE-SWAS) и эпилептическая энцефалопатия в сочетании со спайк-волновой активностью во сне (англ. epileptic encephalopathy in combination with spike-and-wave activation in sleep, EE-SWAS), эпилептический синдром, связанный с фебрильной инфекцией (англ. febrile infection-related epilepsy syndrome, FIRES) и гемиконвульсивно-гемиплегический эпилептический синдром (англ. hemiconvulsion-hemiplegiaepilepsy syndrome, HHE).
EMAtS (ранее именовался «синдромом Дуза») встречается примерно у 1 из 10 тыс. детей и составляет около 2% детских эпилепсий. Полный набор клинических и ЭЭГ-признаков на ранних стадиях может отсутствовать, дополняясь со временем. У пациентов с EMAtS часто наблюдается застой или регресс в развитии на фоне трудно контролируемых приступов с улучшением по мере улучшения контроля над приступами [26][28][29]. Синдром Леннокса–Гасто представляет собой DEE различной этиологии. Он возникает в результате высокочастотной синхронизированной активности в билатерально распределенных сетях мозга. Синдром характеризуется следующими проявлениями:
Авторы классификации обращают внимание, что неверно использовать термин «синдром Леннокса-Гасто» для описания любой тяжелой эпилепсии с ранним началом и трудноконтролируемыми приступами, приводящими к падениям пациента. Такой подход не позволяет распознать специфические особенности синдрома Леннокса-Гасто и отличить его от имеющего лучший исход EMAtS, а также многих других тяжелых эпилепсий, возникающих в детском возрасте. Полный набор клинических и ЭЭГ-признаков на ранних стадиях заболевания часто отсутствует, и для его появления требуется время. Маленькие дети с характерными типами приступов, но без полного набора признаков нуждаются в тщательном наблюдении для выявления трансформации в синдром Леннокса-Гасто. В частности, в данный синдром часто эволюционирует ряд тяжелых синдромов эпилепсии младенчества [26].
DEE-SWAS и ЕЕ-SWAS имеют сходные клинические проявления и характеризуются различными сочетаниями регресса в когнитивной, речевой, поведенческой и двигательной сферах, имеющего место в течение нескольких недель после спайк-волновой активности во сне. В Классификации эпилептических синдромов ILAE 2022 г. они сгруппированы вместе, т.к. имеют схожий исход, включая слуховую агнозию, общий регресс поведенческих и двигательных навыков и негативный миоклонус. DEE-SWAS и EE-SWAS заменяют синдромы, ранее поименованные как «эпилептическая энцефалопатия с продолженной спайк-волновой активностью во сне», «атипичная доброкачественная парциальная эпилепсия (синдром псевдо-Леннокса)». Однако, поскольку синдром Ландау–Клеффнера представляет собой специфический подтип EE-SWAS, при котором регрессия затрагивает в основном речевую функцию с приобретенной слуховой агнозией, это название в новой классификации сохранено (рис. 5).
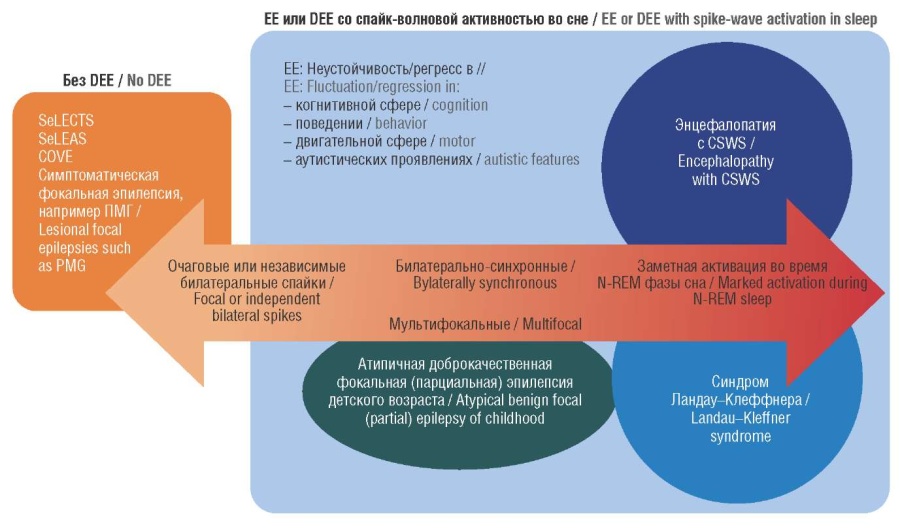
Рисунок 5. Классификация эпилептических синдромов ILAE 2022 г.: эволюционные и/или эпилептические энцефалопатии (англ. developmental and/or epileptic encephalopathy, DEE) или эпилептическая энцефалопатия (англ. epileptic encephalopathy, EE) детского возраста в сочетании со спайк-волновой активностью во сне (англ. spike-and-wave activation in sleep, SWAS) (адаптировано из [26]). На рисунке приведены и более не применяемые названия: «эпилептическая энцефалопатия с продолженной спайкволновой активностью во сне» и «атипичная доброкачественная фокальная (парциальная) эпилепсия». Специфические фокальные эпилептические синдромы, такие как самокупирующаяся эпилепсия с центрально-темпоральными спайками (англ. self-limited epilepsy with centrotemporal spikes, SeLECTS) и самокупирующаяся эпилепсия с вегетативными приступами (англ. self-limited epilepsy with autonomic seizures, SeLEAS) или другие структурные фокальные эпилепсии, могут эволюционировать в EE-SWAS либо транзиторно, либо в течение длительного периода. CSWS (англ. continued spike-wave activity during sleep) – продолженная спайк-волновая активность во время сна; N-REM (англ. non-rapid eye movements) – небыстрые движения глаз; COVE (англ. сhildhood occipital visual epilepsy) – детская затылочная зрительная эпилепсия; ПМГ – полимикрогирия
Figure 5. The 2022 ILAE Classification of Epilepcy Syndromes: developmental and/or epileptic encephalopathies (DEE) or epileptic encephalopathy (EE) of childhood in combination with spike-and-wave activation in sleep (SWAS) (adapted from [26]). No longer used names are also shown: “epileptic encephalopathy with continued spike-wave activity during sleep” and “atypical benign focal (partial) epilepsy.” Specific focal epilepsy syndromes such as self-limited epilepsy with centrotemporal spikes (SeLECTS) and self-limited epilepsy with autonomic seizures (SeLEAS) or other structural focal epilepsies may evolve into EE-SWAS either transiently or during long-term period. CSWS – continued spike-wave activity during sleep; N-REM – non-rapid eye movements; COVE – сhildhood occipital visual epilepsy; PMG – polymicrogyria
Эпилептический синдром, связанный с фебрильной инфекцией (FIRES), ранее также именовавшийся как «острый энцефалит с повторяющимися рефрактерными парциальными приступами» или «злокачественная (разрушительная) эпилептическая энцефалопатия у детей школьного возраста», является одной из причин вновь возникающего рефрактерного эпилептического статуса, который встречается преимущественно у детей и подростков. Этому предшествует лихорадка инфекционного происхождения в течение от 24 ч до 2 нед до его возникновения. Острая фаза, во время которой тяжесть приступов очень велика, длится от 1 до 12 нед и характеризуется высокой вероятностью летального исхода. Далее следует хроническая фаза, когда у большинства выживших остаются фармакорезистентная мультифокальная эпилепсия, умственная отсталость различной степени или трудности в обучении. Причина FIRES неизвестна, но все больше данных свидетельствуют о гетерогенной этиологии, приводящей к молниеносному нейровоспалению, не опосредованному антителами [26][30–32].
Гемиконвульсивно-гемиплегический эпилептический синдром (HHE) является редким следствием фокального моторного эпилептического статуса в младенчестве и раннем детстве. Инициирующим событием для развития данного эпилептического синдрома является фокальный клонический эпилептический статус, обычно возникающий на фоне повышения температуры у детей в возрасте до 4 лет. При нейровизуализации во время эпилептического статуса имеет место односторонний отек пораженного полушария. За острой фазой следует атрофия полушария с последующим появлением фокальных фармакорезистентных приступов. В результате у большинства пациентов возникает постоянный неврологический дефицит. Этиология и лежащие в основе патогенеза HHE механизмы остаются невыясненными [26][33][34].
Синдромы эпилепсии, возникающие в разном возрасте, в Классификации эпилептических синдромов ILAE 2022 г. делятся на следующие группы (рис. 6) [15]:

Рисунок 6. Классификация эпилептических синдромов ILAE 2022 г.: синдромы эпилепсии с началом в разном возрасте (адаптировано из [15])
Figure 6. The 2022 ILAE Classification of Epilepsy Syndromes: epilepsy syndromes with onset at a variable age (adapted from [15])
Авторы классификации эпилептических синдромов с началом в разном возрасте отмечают, что в будущем могут быть выявлены и добавлены в нее другие синдромы [15].
Относящаяся к синдромам генерализованной эпилепсии полигенной этиологии IGE, которую имеют до 20% всех больных эпилепсией, включает JME, JAE, GTCA и в рамках Классификации эпилептических приступов ILAE 2022 г. выделена отдельно [35].
Самокупирующиеся фокальные эпилепсии (SeLFE) составляют до 25% всех детских эпилепсий и преимущественно возникают в детском возрасте, но синдромы COVE и POLE могут начинаться в разном возрасте. Так, для COVE описано начало в возрасте до 19 лет. Хотя при этих синдромах ожидается ремиссия, она может наступить не у всех пациентов. COVE характеризуется частыми короткими фокальными осознанными сенсорными приступами со зрительными явлениями во время бодрствования, за которыми часто следует головная боль. Для ЭЭГ характерны интериктальные затылочные острые или спайк-волны, наблюдаемые в основном во сне. Ремиссия наступает у 50–80% пациентов в течение 2–7 лет после начала заболевания с применением или без применения АЭП. POLE чаще встречается у лиц женского пола, характеризуется фотоиндуцированными фокальными осознанными сенсорными приступами со зрительными явлениями. ЭЭГ отличается интериктальными затылочными спайками или спайк-волнами, появлению которых способствует закрывание глаз и фотостимуляция. Также может иметь место генерализованная спайк-волновая активность [15].
FMTLE – распространенный синдром фокальной эпилепсии со сложным типом наследования, обычно с началом в подростковом или взрослом возрасте. Как правило, FMTLE связан с фокальными осознанными приступами с симптоматикой, относящейся к мезиальной височной доле, особенно с выраженным дежавю.
Пациенты имеют нормальную МРТ и хорошо реагируют на лечение АЭП. Также были описаны некоторые семьи с клинически гетерогенной формой FMTLE, включающей предшествующие фебрильные судороги, МРТ-признаки атрофии гиппокампа и менее благоприятный ответ на АЭП [15][36][37].
EAF представляет собой синдром фокальной эпилепсии, который проявляется в подростковом или взрослом возрасте без какого-либо предшествующего анамнеза и характеризуется фокальными осознанными приступами со слуховыми симптомами и/или рецептивной афазией. В редких случаях у пациентов могут возникать фокальные или билатеральные тонико-клонические припадки. У некоторых больных приступы провоцируются специфическими звуками. EAF ранее именовался как «аутосомно-доминантная латеральная височная эпилепсия» и «аутосомно-доминантная парциальная эпилепсия со слуховыми особенностями». Он может возникать как синдром семейной фокальной эпилепсии, семейный EAF (aнгл. family EAF, FEAF), который может наследоваться по аутосомно-доминантному типу (англ. autosomal dominant EAF, ADEAF) со сниженной пенетрантностью [15].
MTLE-HS – часто встречающаяся фокальная эпилепсия у взрослых, но она может дебютировать и в детском возрасте. Хотя к склерозу гиппокампа способны привести многие факторы, включая генетические, структурно-генетические и иммунные патологии, диагноз MTLE-HS возможно поставить только после подтверждения наличия гиппокампального склероза как причины эпилепсии методами нейровизуализации. MTLE-HS часто резистентен к терапии АЭП, однако хирургическое лечение может привести к полной ремиссии [15].
SHE характеризуется кластерами возникающих во время сна моторных приступов с внезапным началом и прекращением, продолжительностью менее 2 мин, с сохранением сознания и стереотипным гиперкинетическим или асимметричным дистоническим/тоническим двигательным паттерном. SHE может быть устойчивым к терапии АЭП. Авторы Классификации эпилептических синдромов ILAE 2022 г. подчеркивают, что этот синдром может называться либо «гиперкинетической эпилепсией, связанной со сном», либо «гипермоторной эпилепсией, связанной со сном», поскольку у одних пациентов могут быть только гиперкинетические приступы, а у других – фокальные моторные приступы с тоническим/дистоническим компонентом [9][15].
FFEVF представляет собой аутосомно-доминантный синдром семейной фокальной эпилепсии с неполной пенетрантностью, характеризующийся фокальными приступами, которые возникают из разных областей коры (чаще всего лобных или височных) у разных членов семьи с различной степенью тяжести, но у каждого пациента в семье есть одноочаговый тип приступа. Этот синдром ранее был поименован как «семейная парциальная эпилепсия с вариабельными очагами» и «аутосомно-доминантная парциальная эпилепсия с вариабельными очагами». Этиология включает генетические и структурные причины. АЭП в большинстве случаев показывают высокую эффективность. У пациентов с фокальной корковой дисплазией и фармакорезистентными приступами к полной ремиссии может привести хирургическое вмешательство [15][38].
К эпилептическим синдромам с эволюционной и/или эпилептической энцефалопатией или с прогрессирующим ухудшением неврологического статуса авторами классификации отнесен FIRES, который, однако, редко возникает во взрослом возрасте.
Синдром Расмуссена, ранее известный также как «энцефалит Кожевникова–Расмуссена», «энцефалит Расмуссена», характеризуется прогрессирующей атрофией полушарий, верифицируемой методами нейровизуализации. Причина возникновения синдрома Расмуссена остается не установленной, вызывающие заболевание антитела не выявлены. В спинномозговой жидкости могут обнаруживаться умеренный плеоцитоз, слегка повышенный уровень белка и олигоклональные полосы. У пациентов отмечаются фокальные приступы (обычно моторные), частота и тяжесть которых со временем прогрессируют. Развивается прогрессирующий контралатеральный гемипарез. Диагноз ставится на основании характерной клинической картины и данных нейровизуализации. В центральной нервной системе выявляются многоочаговое корковое воспаление, гибель нейронов и глиоз, ограниченный одним полушарием [31].
Синдром PME встречается редко и вызывается гетерогенной группой генетических нарушений. Диагноз PME ставится при наличии миоклонуса, прогрессирующих двигательных и когнитивных нарушений, сенсорных и мозжечковых симптомов и аномального фонового замедления на ЭЭГ у пациента без нарушений развития и когнитивной функции в анамнезе. Общим признаком PME является фотосенсибилизация. PME может быть с аутосомно-рецессивным наследованием (в большинстве случаев), но может и возникать спорадически. Распространенность синдрома более высока в изолированных регионах или в культурах, где одобряются близкородственные браки. Таким образом, место рождения и этническое происхождение пациента являются важными данными для диагностики PME [15][39].
EwRIS — редкий комбинированный синдром генерализованной и фокальной эпилепсии, который характеризуется инициируемыми чтением рефлекторными миоклоническими приступами с поражением орофациальных мышц. Если чтение продолжается, могут возникнуть генерализованные тонико-клонические приступы. Таким образом, тщательный сбор анамнеза имеет решающее значение для постановки диагноза, равно как и осведомленность об этом синдроме, поскольку выявление симптомов в зависимости от задачи может привести к неправильной диагностике как EwRIS, так и тиков или заикания. Приступы вызываются главным образом чтением, но также могут провоцироваться и другими видами активности, связанными с речевой функцией. Прогноз благоприятен, поскольку спонтанные припадки маловероятны, приступы реагируют на лечение и их можно избежать за счет уменьшения воздействия провоцирующего раздражителя. У большинства пациентов судороги требуют длительного лечения, хотя у некоторых со временем может наступить ремиссия [15].
Ранее в контексте классификации эпилепсии термин «идиопатический» использовался для описания расстройств, которым ничего не предшествовало и для которых не было установлено никакой другой причины, кроме возможной наследственной предрасположенности [40][41]. В ходе подготовки Классификации эпилепсии ILAE 2017 г. было признано, что термин «генетический» более точен, чем «идиопатический», однако вместе с тем термин «идиопатическая генерализованная эпилепсия» по-прежнему имеет клиническое значение [9–11][35]. Согласно Классификации эпилептических синдромов ILAE 2022 г. идиопатическая генерализованная эпилепсия (англ. idiopathic generalized epilepsy, IGE) включает четыре синдрома: детская абсансная эпилепсия (CAE), юношеская абсансная эпилепсия (JAE), юношеская миоклоническая эпилепсия (JME) и эпилепсия с изолированными генерализованными тонико-клоническими приступами (GTCA) [35]. Синдромы IGE манифестируют в возрасте от 3 до 25 лет, редко – в более старшем возрасте, но не свыше 40 лет. У 23– 43% больных эпилепсией в детском и подростковом возрасте наблюдается генерализованная эпилепсия, 53–58% из них имеют один из четырех вышеперечисленных синдромов IGE.
В Классификации эпилепсии ILAE 2017 г. предлагается использовать термин «генетические генерализованные эпилепсии» (англ. genetic generalized epilepsies, GGE) для широкой группы эпилепсий с генерализованными типами приступов и генерализованными спайк-волнами, предположительно генетической этиологии, установленной на основании наличия схожих проявлений у близнецов, а также членов семей [9–11][35]. Признавая, что группа GGE включает множество как распространенных, так и редко встречающихся генетических синдромов генерализованной эпилепсии, Рабочая группа ILAE отнесла IGE к отдельной подгруппе GGE по следующим причинам [35]:
На рисунке 7 показано, как IGE входят в большую группу GGE. Здесь отражено, что различие между четырьмя синдромами IGE не всегда четкие, поскольку имеет место схожесть клинических проявлений. Кроме того, существует пересечение между IGE и не-IGE GGE, о чем свидетельствует более высокая частота синдромов IGE у родственников больных эпилепсией с миоклонией век, эпилепсией с миоклоническими абсансами, миоклонической эпилепсией младенчества, эпилепсией с миоклоническими атоническими приступами и генетической эпилепсией с фебрильными судорогами плюс (GEFS+) [23][35][42][43].

Рисунок 7. Классификация эпилептических синдромов ILAE 2022 г.: генетические генерализованные эпилепсии, включающие идиопатические генерализованные эпилепсии (адаптировано из [35])
Figure 7. The 2022 ILAE Classification of Epilepsy Syndromes: genetic generalized epilepsies including idiopathic generalized epilepsies (adapted from [35])
Помимо IGE к группе GGE относятся генерализованные типы приступов, которые не соответствуют критериям какого-либо синдрома, и менее распространенные синдромы генерализованной эпилепсии, также имеющие генетическую основу, развивающиеся в условиях нормального интеллекта либо отставания в умственном развитии. Некоторые больные могут иметь эпилептическую энцефалопатию (EE), такую как эпилепсия с миоклонически-атоническими приступами (EMAtS), другие – эволюционную и эпилептическую энцефалопатию (DEE), к которой относятся такие синдромы, как эпилепсия с миоклоническими абсансами и эпилепсия с миоклонией век. Ряд эпилептических синдромов, таких как миоклоническая эпилепсия младенчества, может проявляться генерализованной эпилепсией у ребенка с эволюционной энцефалопатией (т.е. отставанием в умственном развитии) или с нормальным интеллектом. Пациенты с IGE испытывают один или комбинацию следующих типов генерализованных приступов: абсансы, миоклонические, тонико-клонические и миоклонико-тонико-клонические приступы. Генерализованные тонико-клонические судороги могут иметь в качестве ранних проявлений фокальные или асимметричные компоненты, такие как отклонение или версивные движения головы и глаз, а миоклонические судороги могут быть фокальными или асимметричными. Очаговые проявления часто варьируют от приступа к приступу. У части пациентов с IGE имеет место фотосенсибилизация [35].
При IGE на ЭЭГ присутствуют генерализованные разряды спайк-волн, обычно 2,5–5,5 Гц, которые часто возникают на фоне депривации сна, во время сна и при пробуждении. При фотостимуляции у большинства не получающих лечение пациентов с JME и у меньшинства пациентов с CAE и JAE возникает фотопароксизмальный ответ. Нормальная рутинная ЭЭГ не исключает диагноз IGE при наличии убедительных клинических данных.
У пациентов с IGE часто наблюдаются расстройства настроения, тревожность, синдром дефицита внимания и гиперактивности (СДВГ) и трудности в обучении. IGE также коррелирует с более низкими долгосрочными социальными исходами, включая снижение успеваемости, повышенный риск незапланированной беременности, психические, эмоциональные и поведенческие проблемы, снижение социального взаимодействия с друзьями. У небольшой части пациентов с IGE выявлены моногенные причины. Примеры включают мутации в нескольких генах – субъединицах рецептора гамма-аминомасляной кислоты (например, GABRG2, GABRA1) и гене, кодирующем переносчик глюкозы 1-го типа (SLC2A1). Встречаются как унаследованные варианты, так и варианты de novo. В первом случае семейный анамнез может показывать неполную пенетрантность (при этом здоровые родственники являются носителями патогенного варианта), во втором случае больные эпилепсией среди членов семьи отсутствуют.
Хотя семейный анамнез эпилепсии, связанной с генерализованными приступами, подтверждает это, чаще всего пациенты с IGE не имеют семейного анамнеза эпилепсии. Это объясняется либо мутацией de novo, либо сложным наследованием. Таким образом, термин «генетический» относится к предрасполагающей причине, а не к наследованию самой болезни.
Существует достаточное количество больных GGE, имеющих генерализованные спайк-волны на ЭЭГ и генерализованные приступы, но клинически не соответствующих ни одному синдрому IGE (например, пациенты с миоклонической эпилепсией младенчества, эпилепсией с миоклонией век, эпилепсией с миоклоническими абсансами и эпилепсией с миоклонически-атоническими приступами или вообще с проявлениями, не соответствующими ни одному синдрому). Такие больные должны быть классифицированы как пациенты с GGE без специфического эпилептического синдрома.
Детская абсансная эпилепсия (CAE) возникает у нормального в других отношениях ребенка. Синдром характеризуется наличием ежедневных абсансов, при этом на ЭЭГ в начале приступа регистрируется генерализованная спайк-волновая активность частотой 2,5–4 Гц. Абсансы провоцируются гипервентиляцией. Неврологический статус без патологических проявлений, развитие и когнитивная сфера без отклонений от нормы. У детей с абсансной эпилепсией может иметь место СДВГ и могут появляться трудности с обучением. Приступы кратковременны, но могут возникать кластерами. Эпилепсия переходит в стадию ремиссии у 60% детей чаще всего в течение 2 лет после возникновения или в раннем подростковом возрасте [35][44].
Юношеская абсансная эпилепсия (JAE) характеризуется абсансами у нормального в других отношениях подростка, которые обычно возникают реже чем один раз в день при отсутствии лечения и проявляются на ЭЭГ генерализованной спайк-волновой активностью частотой 3–5,5 Гц. Генерализованные тонико-клонические приступы наблюдаются более чем в 90% случаев, чаще всего они начинаются вскоре после начала абсансов. Неврологический статус без патологических проявлений, развитие и когнитивная сфера обычно без особенностей, хотя могут иметь место СДВГ и трудности в обучении. JAE хорошо контролируется АЭП, но может потребоваться пожизненная терапия [35][45] .
Юношеская миоклоническая эпилепсия (JME) является наиболее частым синдромом IGE с дебютом в подростковом и взрослом возрасте. JME характеризуется миоклоническими и генерализованными тонико-клоническими судорогами у нормальных в других отношениях подростков и взрослых. Миоклонические приступы обычно возникают вскоре после пробуждения и при утомлении. Депривация сна является важным провоцирующим фактором. На ЭЭГ регистрируется генерализованная спайк-волновая и полиспайк-волновая активность частотой 3–5,5 Гц. Фотосенситивность при JME – распространенное явление, возникающее у 90% больных при использовании фотостимуляции. Пациентам с JME часто требуется пожизненная терапия [35][46–48].
Эпилепсия с изолированными генерализованными тонико-клоническими приступами (GTCA) является довольно часто встречающимся синдромом IGE. У части пациентов наблюдаются генерализованные тонико-клонические приступы, возникающие с различной частотой, которые обычно дебютируют на втором или в начале третьего десятилетия жизни и, как правило, провоцируются депривацией сна. Других типов приступов не отмечается. На ЭЭГ имеет место генерализованная спайк-волновая или полиспайк-волновая активность частотой 3–5,5 Гц. Частота ремиссии низкая, пациентам с GTCA может потребоваться пожизненное лечение [35].
В классификации ILAE прошлых лет и в предложениях ILAE по уточнениям определений и классификаций всегда упоминался ряд хорошо изученных и довольно часто встречающихся эпилептических синдромов. Однако официально утвержденной ILAE синдромальной классификации до недавних пор принято не было. В 2017 г. ILAE представила базовую классификацию типов эпилептических приступов, тогда же эксперты отметили, что следующей логической областью для рассмотрения и оценки должна стать классификация эпилептических синдромов. Была создана Рабочая группа ILAE и началась активная фаза создания классификации. Потребовалось 5 лет упорной работы экспертного сообщества, чтобы разработать, согласовать и утвердить классификацию эпилептических синдромов.
Уникальным преимуществом стала применяемая методология, когда экспертная оценка и согласование проходят с использованием дельфийского метода в несколько итераций с участием экспертов Рабочей группы ILAE, специалистов авторитетных медицинских общественных организаций, широкой общественности, имевшей возможность внести вклад в экспертизу через официальный сайт ILAE, и рецензентов журнала Epilepsia. Таким образом, было обеспечено участие практически всех специалистов здравоохранения, имеющих интересы и экспертный уровень в эпилептологии и смежных направлениях. Однако нет информации об участии в процессе пациентских общественных организаций, которые по установившейся в последние годы практике привлекаются для создания гайдлайнов по ограниченному кругу других нозологий. Но в любом случае описанный формат представляет собой редкий случай организованной совместной работы, когда каждый специалист при желании мог участвовать в создании документа и мнение каждого эксперта влияло на конечный результат.
Важными преимуществами Классификации эпилептических синдромов ILAE 2022 г. является разработанная единая структура описания, включающая эпидемиологию, анамнез, клиническую картину, течение болезни, описание приступов, электроклиническую картину (ЭЭГ), нейровизуализацию, данные генетических исследований, дифференциальную диагностику, а также диагностические критерии в едином формате (обязательные критерии, настораживающие признаки и критерии исключения). Пожалуй, впервые представлена выполненная по единой схеме детальная характеристика каждого из эпилептических синдромов, вошедших в классификацию, которую в переводе на русский язык вы можете найти в Приложениях к этой статье.
Несомненно, позитивным моментом является решение сохранить ряд прежних названий синдромов, являющихся эпонимами, таких как синдром Драве, синдром Расмуссена, синдром Леннокса–Гасто. Те, кто следил за ходом разработки классификации, могли отмечать публичные дискуссии на этот счет, но в итоге было принято решение оставить их, хотя подавляющее большинство других названий-эпонимов было заменено на название по доминирующему типу приступов с целью максимально точно отразить их клиническую картину. Основанием сохранения прежних названий для вышеперечисленных синдромов послужило то, что это устоявшиеся и широко применяющиеся в рутинной клинической практике наименования. Также в настоящее время проходят начатые несколько лет назад клинические исследования, ряд препаратов в инструкции по медицинскому применению имеют данные синдромы в составе показаний. Поэтому изменение названия повлекло бы за собой терминологическую путаницу, необходимость внесения изменений в протоколы клинических исследований и инструкции по медицинскому применению, что в итоге ухудшило бы доступ пациентов к терапии.
Новая Классификация эпилептических синдромов ILAE 2022 г. является одной из самых объемных медицинских классификаций. Она занимает в общей сложности 166 страниц в официальном печатном органе ILAE – рецензируемом журнале Epilepsia [12–15][26][35]. Очевидно, что практикующие врачи должны будут постоянно обращаться к первоисточнику, однако быстрый поиск необходимой им информации и в случае обращения будет затруднен, поскольку классификация не представляет собой единый документ, а разделена на шесть разных статей. Между тем объем можно было сократить: так, раздел «Методология» не только подробно описан в отдельной публикации, но и во многом дублирован в каждой из статей.
Сейчас описание одного и того же эпилептического синдрома с разной степенью детализации может присутствовать в нескольких статьях, составляющих Классификацию эпилептических синдромов ILAE 2022 г. Очевидно, что это во многом обусловлено сложностью и многогранностью проблематики эпилепсии, тем не менее это не идет на пользу классификации, поскольку в результате затруднен быстрый поиск в условиях ограниченного временного ресурса практикующего врача. Также могут возникать разночтения в трактовке и невольные заблуждения в отношении того, как классифицировать такие эпилептические синдромы. Кроме этого, встречаются небольшие различия в терминологии. Например, в статье S. Zuberi et al. [14] используется термин «эволюционная и эпилептическая энцефалопатия» (англ. developmental and epileptic encephalopathies), а в работе N. Specchio et al. [26] преимущественно встречается термин «эволюционная и/или эпилептическая энцефалопатия», (англ. developmental and/or epileptic encephalopathies). В статьях K. Riney et al. [15], E. Hirsch et al. [35] и E.C. Wirrell et al. [13] используются оба варианта, при этом аббревиатура одна и та же (DEE). Работа K. Riney et al. [15] содержит пояснение: «Термин “эволюционная энцефалопатия” применяется, когда возникает состояние, проявляющееся когнитивными, неврологическими или психическими нарушениями, застоем или регрессией, непосредственно обусловленными основной этиологией.
Напротив, термин “эпилептическая энцефалопатия” присутствует, когда энцефалопатия вызвана эпилептической активностью. Термин “эволюционная и эпилептическая энцефалопатия” (DEE) используется, когда оба фактора способствуют состоянию пациента». Повидимому, такие моменты обусловлены тем, что над различными составляющими классификации работали разные коллективы авторов.
Иллюстрирующие классификацию схемы не всегда легки для восприятия, также авторы не всегда дают подробные пояснения – как, например, E. Hirsch et al. [35] представили пояснения к блок-схеме классификации генетических генерализованных эпилепсий, включающих идиопатические генерализованные эпилепсии (см. рис. 7, описание приведено в тексте статьи). В ряде других случаев не вполне понятно, что означает тот или иной цвет или градиент на схеме, несет ли это какой-либо смысл или сделано лишь для лучшего визуального восприятия. Кроме того, на блок-схеме, иллюстрирующей DEE или EE детского возраста в сочетании со спайк-волновой активностью во сне [26], присутствуют старые наименования эпилептических синдромов, от которых уже отказались в Классификации эпилептических синдромов ILAE 2022 г., что также может запутать практикующих врачей (см. рис. 5).
Составители классификации не всегда четко придерживаются разработанной единой схемы описания эпилептических синдромов. В качестве одного из примеров: в описании эпилептических синдромов с началом в детском возрасте для части из них отдельным пунктом в таблицах приведен долгосрочный прогноз, в других случаях эта информация не дается. Другим примером являются начинающиеся в разном возрасте эпилептические синдромы с эволюционной и/или эпилептической энцефалопатией и эпилептические синдромы с прогрессирующим ухудшением неврологического статуса (болезни Унверрихта–Лундборга, Лафора, нейрональный цероидный липофусциноз), описание которых дается сплошным текстом без структурирования по вышеописанному шаблону.
Наконец, авторы декларируют стремление использовать четкую терминологию, которую можно было бы легко перевести на разные языки для удобства применения международным сообществом [13]. Однако эта задача была решена не в полной мере. На английском языке многие аббревиатуры звучат весьма благозвучно, но перевод на русский язык в ряде случаев затруднен. Например, термин developmental and/or epileptic encephalopathy (DEE) в данной статье переведен для краткости «эволюционная и эпилептическая энцефалопатия» или «эволюционная и/или эпилептическая энцефалопатия» в зависимости от контекста оригинальных публикаций, хотя более точным является перевод «энцефалопатия, связанная с развитием и/или эпилепсией», а аббревиатура на русском неудобна для повседневного произношения, поэтому параллельно используется и аббревиатура на английском. Как выход можно использовать англоязычные аббревиатуры (в статье преимущественно применяется такой подход), но это потребует привыкания практикующих врачей. Другой пример: selflimited epilepsy with centrotemporal spikes с запоминающейся аббревиатурой SeLECTS переводится как «самокупирующаяся эпилепсия с центрально-темпоральными спайками», и аббревиатура на русском неизбежно теряет в красоте и запоминаемости. Также термин «самокупирующаяся эпилепсия» может быть ошибочно трактован как эпилепсия, не требующая лечения, которая в будущем исчезнет сама по себе. Поэтому в русскоязычном сообществе специалистов, занимающихся проблемой эпилепсии, достаточно широко распространен термин «возраст-зависимая эпилепсия». Но здесь с целью применения терминологии, максимально близкой к оригинальной терминологии Классификации эпилептических синдромов ILAE 2022 г. приводится термин «самокупирующаяся эпилепсия».
Классификация эпилептических синдромов ILAE 2022 г. является итогом многолетних усилий большого количества экспертов и примером открытого международного сотрудничества специалистов в области эпилепсии и смежных направлений. Сложность и многогранность проблемы обусловливают возможную критику новой классификации со стороны профессионального сообщества, которая наверняка будет иметь место. Тем не менее выполненная работа по определению и классификации эпилептических синдромов – важный шаг вперед и отправная точка как для улучшения понимания их этиопатогенеза, так и для дальнейшего совершенствования организации работы практикующих специалистов, которые занимаются проблемой эпилепсии, во всем мире.
В какой мере Классификация эпилептических синдромов ILAE 2022 г. получит признание со стороны практикующих специалистов и насколько широко она будет внедрена в клиническую практику – покажет время.
Статья и Приложения к статье размещены онлайн на интернет-сайте журнала «Эпилепсия и пароксизмальные состояния»: https://epilepsia.su. Считайте QR-код камерой смартфона.


Приложение A1. Эпилептические синдромы с началом в неонатальном периоде и младенчестве

Приложение А2. Диагностические критерии эпилептических синдромов с началом вVнеонатальном периоде и младенчестве

Приложение В1. Эпилептические синдромы с началом в детском возрасте

Приложение В2. Диагностические критерии эпилептических синдромов с началом в детском возрасте

Приложение В3. Эпилептические синдромы с началом в детском возрасте: сравнение названий

Приложение С1. Эпилептические синдромы с началом в разном возрасте

Приложение С2. Диагностические критерии эпилептических синдромов с началом в разном возрасте

Приложение С3. Отличительные особенности связанной со сном гипермоторной (гиперкинетической) эпилепсии, семейной мезиальной височной эпилепсии, семейной фокальной эпилепсии с вариабельными очагами и эпилепсии со слуховыми приступами

Приложение D1. Синдромы идиопатической генерализованной эпилепсии

Приложение D2. Диагностические критерии синдромов идиопатической генерализованной эпилепсии

Приложение D3. Отличительные особенности детской абсансной эпилепсии, юношеской абсансной эпилепсии, юношеской миоклонической эпилепсии и эпилепсии с изолированными генерализованными тонико-клоническими приступами
1. Карлов В.А. Эпилепсия у детей и взрослых женщин и мужчин. Руководство для врачей. 2-е изд. М.: БИНОМ; 2019: 806 с.
2. Hauser W.A., Annegers J.F., Kurland L.T. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935–1984. Epilepsia. 1993; 34 (3): 453–68. https://doi.org/10.1111/j.1528-1157.1993.tb02586.x.
3. Epilepsy: a public health imperatives. Geneva: World Health Organization, 2019. URL: https://www.who.int/publications-detail-redirect/epilepsy-a-public-health-imperative (дата обращения 07.06.2022).
4. Карлов В.А. Российская Противоэпилептическая Лига сегодня: вызов времени. Эпилепсия и пароксизмальные состояния. 2020; 12 (1): 5.
5. Карлов В.А. Что удалось и чего не удалось достичь в 2020 году. Эпилепсия и пароксизмальные состояния. 2021; 13 (1): 5.
6. Карлов В.А. Российская Противоэпилептическая Лига: ответ на вызов времени. Эпилепсия и пароксизмальные состояния. 2021; 13 (1S): S88–90. https://doi.org/10.17749/2077-8333/epi.par.con.2021.085.
7. Political declaration of the third high-level meeting of the General Assembly on the prevention and control of noncommunicable diseases. Geneva: World Health Organization; 2022. URL: https://apps.who.int/gb/ebwha/pdf_files/EB150/B150_7-en.pdf (дата обращения 07.06.2022).
8. Межсекторальный глобальный план действий по борьбе с эпилепсией и другими неврологическими расстройствами 2022–2031 гг. Всемирная организация здравоохранения; 2021. URL: https://cdn.who.int/media/docs/default-source/brain-health/first-draft-action-plan-on-epilepsy-and-other-neurological-disorders-180621-ru83acba3d76f947f98992fcd956c832e5.pdf?sfvrsn=16474e26_24&download=true (дата обращения 07.06.2022).
9. Fisher R.S., Cross J.H., French J.A., et al. Operational classification of seizure types by the International League Against Epilepsy: Position Paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017; 58 (4): 522–30. https://doi.org/10.1111/epi.13670.
10. Scheffer I.E., Berkovic S., Capovilla G., et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017; 58 (4): 512–21. https://doi.org/10.1111/epi.13709.
11. Авакян Г.Н., Блинов Д.В., Лебедева А.В. и др. Классификация эпилепсии Международной Противоэпилептической Лиги: пересмотр и обновление 2017 года. Эпилепсия и пароксизмальные состояния. 2017; 9 (1): 6–25. https://doi.org/10.17749/2077-8333.2017.9.1.006-025.
12. Wirrell E., Tinuper P., Perucca E., et al. Introduction to the epilepsy syndrome papers. Epilepsia. 2022; 63 (6): 1330–2. https://doi.org/10.1111/epi.17262.
13. Wirrell E.C., Nabbout R., Scheffer I.E., et al. Methodology for classification and definition of epilepsy syndromes with list of syndromes: report of the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022; 63 (6): 1333–48. https://doi.org/10.1111/epi.17237.
14. Zuberi S.M., Wirrell E., Yozawitz E., et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022; 63 (6): 1349–97. https://doi.org/10.1111/epi.17239.
15. Riney K., Bogacz A., Somerville E., et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset at a variable age: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022; 63 (6): 1443–74. https://doi.org/10.1111/epi.17240.
16. Linstone H.A., Turoff M. (Eds.) The Delphi method: techniques and applications. URL: https://web.njit.edu/~turoff/pubs/delphibook/delphibook.pdf (дата обращения 07.06.2022).
17. Symonds J.D., Elliott K.S., Shetty J., et al. Early childhood epilepsies: epidemiology, classification, aetiology, and socio-economic determinants. Brain. 2021; 144 (9): 2879–91. https://doi.org/10.1093/brain/awab162.
18. Wirrell E., Wong-Kisiel L., Mandrekar J., Nickels K. Predictors and course of medically intractable epilepsy in young children presenting before 36 months of age: a retrospective, population-based study. Epilepsia. 2012; 53 (9): 1563–9. https://doi.org/10.1111/j.1528-1167.2012.03562.x.
19. Moseley B.D., Wirrell E.C., Wong-Kisiel L.C., Nickels K. Early onset epilepsy is associated with increased mortality: a population-based study. Epilepsy Res. 2013; 105 (3): 410–14. https://doi.org/10.1016/j.eplepsyres.2013.03.002.
20. Berg A.T., Langfitt J.T., Testa F.M., et al. Global cognitive function in children with epilepsy: a community-based study. Epilepsia. 2008; 49 (4): 608–14. https://doi.org/10.1111/j.1528-1167.2007.01461.x.
21. Berg A.T., Zelko F.A., Levy S.R., Testa F.M. Age at onset of epilepsy, pharmacoresistance, and cognitive outcomes: a prospective cohort study. Neurology. 2012; 79 (13): 1384–91. https://doi.org/10.1212/WNL.0b013e31826c1b55.
22. Wilson S.J., Micallef S., Henderson A., et al. Developmental outcomes of childhood-onset temporal lobe epilepsy: a community-based study. Epilepsia. 2012; 53 (9): 1587–96. https://doi.org/10.1111/j.1528-1167.2012.03632.x.
23. Шарков А.А. Генетическая эпилепсия с фебрильными судорогами плюс (GEFS+). Эпилепсия и пароксизмальные состояния. 2020; 12 (1S): S50–6. https://doi.org/10.17749/2077-8333.2020.12.1S.S50-S56.
24. Camfield P., Camfield C. Incidence, prevalence and aetiology of seizures and epilepsy in children. Epileptic Disord. 2015; 17 (2) 117–23. https://doi.org/10.1684/epd.2015.0736.
25. Wirrell E.C., Grossardt B.R., Wong-Kisiel L.C., Nickels K.C. Incidence and classification of new-onset epilepsy and epilepsy syndromes in children in Olmsted County, Minnesota from 1980 to 2004: a population-based study. Epilepsy Res. 2011; 95 (1–2): 110–8. https://doi.org/10.1016/j.eplepsyres.2011.03.009.
26. Specchio N., Wirrell E.C., Scheffer I.E., et al. International League Against Epilepsy classification and definition of epilepsy syndromes with onset in childhood: position paper by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022; 63 (6): 1398–442. https://doi.org/10.1111/epi.17241.
27. Verrotti A., D’Alonzo R., Rinaldi V.E., et al. Childhood absence epilepsy and benign epilepsy with centro-temporal spikes: a narrative review analysis. World J Pediatr. 2017; 13 (2): 106–11. https://doi.org/10.1007/s12519-017-0006-9.
28. Kelley S.A., Kossoff E.H. Doose syndrome (myoclonic–astatic epilepsy): 40 years of progress. Dev Med Child Neurol. 2010; 52 (11): 988–93. https://doi.org/10.1111/j.1469-8749.2010.03744.x.
29. Doose H. Myoclonic-astatic epilepsy. Epilepsy Res Suppl. 1992; 6: 163–8.
30. Hirsch L.J., Gaspard N., van Baalen A., et al. Proposed consensus definitions for new-onset refractory status epilepticus (NORSE), febrile infection-related epilepsy syndrome (FIRES), and related conditions. Epilepsia. 2018; 59 (4): 739–44. https://doi.org/10.1111/epi.14016.
31. Payne E.T., Koh S., Wirrell E.C. Extinguishing febrile infection-related epilepsy syndrome: pipe dream or reality? Semin Neurol. 2020; 40 (2): 263–72. https://doi.org/10.1055/s-0040-1708503.
32. Specchio N., Pietrafusa N. New-onset refractory status epilepticus and febrile infection-related epilepsy syndrome. Dev Med Child Neurol. 2020; 62 (8): 897–905. https://doi.org/10.1111/dmcn.14553.
33. Gastaut H., Poirier F., Payan H., et al. H.H.E. syndrome; hemiconvulsions, hemiplegia, epilepsy. Epilepsia. 1960; 1: 418–47. https://doi.org/10.1111/j.1528-1157.1959.tb04278.x.
34. Айвазян С.О. Коморбидность при синдроме гемиконвульсий-гемиплегии-эпилепсии у детей (описание четырех клинических случаев). Эпилепсия и пароксизмальные состояния. 2018; 10 (1): 73–9. https://doi.org/10.17749/2077-8333.2018.10.1.073-079.
35. Hirsch E., French J., Scheffer I.E., et al. ILAE definition of the Idiopathic Generalized Epilepsy Syndromes: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022; 63 (6): 1475–99. https://doi.org/10.1111/epi.17236.
36. Cendes F., Lopes-Cendes I., Andermann E., Andermann F. Familial temporal lobe epilepsy: a clinically heterogeneous syndrome. Neurology. 1998; 50 (2): 554–7. https://doi.org/10.1212/WNL.50.2.554.
37. Kobayashi E., Lopes-Cendes I., Guerreiro C.A., et al. Seizure outcome and hippocampal atrophy in familial mesial temporal lobe epilepsy. Neurology. 2001; 56 (2): 166–72. https://doi.org/10.1212/wnl.56.2.166.
38. Berkovic S.F., Serratosa J.M., Phillips H.A., et al. Familial partial epilepsy with variable foci: clinical features and linkage to chromosome 22q12. Epilepsia. 2004; 45 (9): 1054–60. https://doi.org/10.1111/j.0013-9580.2004.30502.x.
39. Bureau M., Genton P., Dravet C., et al. Epileptic syndromes in infancy, childhood and adolescence. 5th ed. John Libbey Eurotext; 2012: 600 pp.
40. Proposal for classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia. 1985; 26 (3): 268–78.
41. Proposal for revised classification of epilepsies and epileptic syndromes. Commission on Classification and Terminology of the International League Against Epilepsy. Epilepsia. 1989; 30 (4): 389–99. https://doi.org/10.1111/j.1528-1157.1989.tb05316.x.
42. Angione K., Eschbach K., Smith G., et al. Genetic testing in a cohort of patients with potential epilepsy with myoclonic-atonic seizures. Epilepsy Res. 2019; 150: 70–7. https://doi.org/10.1016/j.eplepsyres.2019.01.008.
43. Zhang Y.H., Burgess R., Malone J.P., et al. Genetic epilepsy with febrile seizures plus: refining the spectrum. Neurology. 2017; 89 (12): 1210–9. https://doi.org/10.1212/WNL.0000000000004384.
44. Alwadei A.H. Childhood absence epilepsy: electro-clinical manifestations, treatment options, and outcome in a tertiary educational center. Int J Pediatr Adolesc Med. 2022; 9 (2): 131–5. https://doi.org/10.1016/j.ijpam.2021.11.003.
45. Elmali A.D., Auvin S., Bast T., et al. How to diagnose and classify idiopathic (genetic) generalized epilepsies. Epileptic Disord. 2020; 22 (4): 399–420. https://doi.org/10.1684/epd.2020.1192.
46. Turco F., Bonanni E., Milano C., et al. Prolonged epileptic discharges predict seizure recurrence in JME: insights from prolonged ambulatory EEG. Epilepsia. 2021; 62 (5): 1184–92. https://doi.org/10.1111/epi.16875.
47. Pietrafusa N., La Neve A., de Palma L., et al. Juvenile myoclonic epilepsy: long-term prognosis and risk factors. Brain Dev. 2021; 43 (6): 688–97. https://doi.org/10.1016/j.braindev.2021.02.005.
48. Amrutkar C., Riel-Romero R.M. Juvenile myoclonic epilepsy. Treasure Island (FL): StatPearls Publishing; 2021.
Блинов Дмитрий Владиславович – к.м.н., руководитель по научным и медицинским вопросам; врач-невролог
WoS ResearcherID: J-4946-2017;
Scopus Author ID: 7003589812;
РИНЦ SPIN-код: 6317-9833
ул. Садовая-Триумфальная, д. 4/10, Москва 127006
1-е Успенское ш., д. 111, Московская обл., Одинцовский р-н, Лапино 143081

|
1. Приложение A1. Эпилептические синдромы с началом в неонатальном периоде и младенчестве | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(1MB)
|
Метаданные ▾ | |
|
|
2. Приложение А2. Диагностические критерии эпилептических синдромов с началом в неонатальном периоде и младенчестве | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(1MB)
|
Метаданные ▾ | |
|
|
3. Приложение В1. Эпилептические синдромы с началом в детском возрасте | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(1MB)
|
Метаданные ▾ | |
|
|
4. Приложение В2. Диагностические критерии эпилептических синдромов с началом в детском возрасте | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(1004KB)
|
Метаданные ▾ | |
|
|
5. Приложение В3. Эпилептические синдромы с началом в детском возрасте: сравнение названий | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(755KB)
|
Метаданные ▾ | |
|
|
6. Приложение С1. Эпилептические синдромы с началом в разном возрасте | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(986KB)
|
Метаданные ▾ | |
|
|
7. Приложение С2. Диагностические критерии эпилептических синдромов с началом в разном возрасте | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(916KB)
|
Метаданные ▾ | |
|
|
8. Приложение С3. Отличительные особенности связанной со сном гипермоторной (гиперкинетической) эпилепсии, семейной мезиальной височной эпилепсии, семейной фокальной эпилепсии с вариабельными очагами и эпилепсии со слуховыми приступами | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(769KB)
|
Метаданные ▾ | |
|
|
9. Приложение D1. Синдромы идиопатической генерализованной эпилепсии | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(946KB)
|
Метаданные ▾ | |
|
|
10. Приложение D2. Диагностические критерии синдромов идиопатической генерализованной эпилепсии | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(855KB)
|
Метаданные ▾ | |
|
|
11. Приложение D3. Отличительные особенности детской абсансной эпилепсии, юношеской абсансной эпилепсии, юношеской миоклонической эпилепсии и эпилепсии с изолированными генерализованными тонико-клоническими приступами | |
| Тема | ||
| Тип | Исследовательские инструменты | |
Скачать
(798KB)
|
Метаданные ▾ | |
Блинов Д.В. Эпилептические синдромы: определение и классификация Международной Противоэпилептической Лиги 2022 года. Эпилепсия и пароксизмальные состояния. 2022;14(2):101-182. https://doi.org/10.17749/2077-8333/epi.par.con.2022.123
Blinov D.V. Epilepsy syndromes: the 2022 ILAE definition and classification. Epilepsy and paroxysmal conditions. 2022;14(2):101-182. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2022.123

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































