


Содержание
Перейти к:
 Т. В. Кожанова,
С. С. Жилина,
Т. И. Мещерякова,
Е. Г. Лукьянова,
Е. С. Большакова,
С. О. Айвазян,
К. В. Осипова,
П. А. Власов,
А. И. Крапивкин,
Н. Н. Заваденко
Т. В. Кожанова,
С. С. Жилина,
Т. И. Мещерякова,
Е. Г. Лукьянова,
Е. С. Большакова,
С. О. Айвазян,
К. В. Осипова,
П. А. Власов,
А. И. Крапивкин,
Н. Н. Заваденко https://doi.org/10.17749/2077-8333/epi.par.con.2023.150
Перейти к:
В статье представлены клинические наблюдения 6 пациентов с эпилепсией, задержкой психомоторного и речевого развития. При проведении полноэкзомного секвенирования выявлены гетерозиготные варианты нуклеотидной последовательности в гене SPTAN1. Мутации в гене SPTAN1 описаны у пациентов с энцефалопатией развития и эпилептической энцефалопатией 5-го типа (англ. developmental and epileptic encephalopathy 5; ОMIM: 613477). Клинический анамнез, данные электроэнцефалографии и магнитно-резонансной томографии наших пациентов схожи с данными детей с вариантами в гене SPTAN1, ранее описанными в научной литературе. Показано, что варианты в гене SPTAN1, локализованные ближе к С-терминальному региону, ассоциированы с более тяжелым фенотипом, а варианты вблизи N-региона – с более легким течением заболевания без структурных аномалий головного мозга. Однако необходимы дальнейшие исследования для лучшего понимания генотип-фенотипических корреляций при SPTAN1-ассоциированной энцефалопатии.
Кожанова Т.В., Жилина С.С., Мещерякова Т.И., Лукьянова Е.Г., Большакова Е.С., Айвазян С.О., Осипова К.В., Власов П.А., Крапивкин А.И., Заваденко Н.Н. SPTAN1-ассоциированная энцефалопатия развития и эпилептическая энцефалопатия. Эпилепсия и пароксизмальные состояния. 2023;15(3):246–259. https://doi.org/10.17749/2077-8333/epi.par.con.2023.150
Kozhanova T.V., Zhilina S.S., Meshcheryakova T.I., Lukyanova E.G., Bolshakova E.S., Ayvazyan S.O., Osipova K.V., Vlasov P.A., Krapivkin A.I., Zavadenko N.N. SPTAN1-associated developmental and epileptic encephalopathy. Epilepsy and paroxysmal conditions. 2023;15(3):246–259. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2023.150
Энцефалопатия развития и эпилептическая энцефалопатия (ЭРиЭЭ) представляют собой группу неврологических заболеваний, которая характеризуется ранним началом фармакорезистентных приступов, тяжелыми электроэнцефалографическими нарушениями, задержкой развития и интеллектуальным дефицитом [1]. Судороги, начинающиеся на первом году жизни, в т.ч. в неонатальном периоде, могут иметь благоприятное течение, например у детей с доброкачественной семейной неонатальной эпилепсией, простыми фебрильными или острыми симптоматическими судорогами. Однако в некоторых случаях эпилепсия с началом на первом году жизни имеет прогрессирующее течение с выраженными церебральными нарушениями [2][3].
Данная группа включает раннюю младенческую эволюционную и эпилептическую энцефалопатию, эпилепсию младенчества с мигрирующими фокальными приступами, синдром инфантильных эпилептических спазмов и синдром Драве [3]. Основными причинами эпилепсии являются структурные аномалии головного мозга, врожденные нарушения метаболизма и приобретенные повреждения головного мозга. Однако в течение многих лет патогенез ряда случаев ЭРиЭЭ оставался неизвестным. В последнее десятилетие генетические исследования показали, что значительная часть ранних криптогенных ЭРиЭЭ ассоциирована с мутациями в генах, участвующих в нейроонтогенезе [4].
SPTAN1 входит в число генов, варианты нуклеотидной последовательности в которых связаны с энцефалопатией развития и эпилептической энцефалопатией 5-го типа (OMIM: 613477) [5]. Этот ген кодирует спектрин αII, один из гибких субмембранных каркасных белков, участвующих в стабилизации клеточных мембран. Две α- и пять β-субъединиц спектрина были идентифицированы. Данные субъединицы собираются антипараллельно бок о бок в гетеродимеры, образовывая тетрамеры, которые, в свою очередь, интегрируются в цитоскелет мембраны. Поскольку целостность гетеротетрамера необходима для развития нейронных отростков и образования тормозных синапсов [6], патогенные варианты в гене SPTAN1 могут приводить к выраженным неврологическим нарушениям [7].
Однако клинические проявления у проанализированных 34 человек со SPTAN1-ассоциированными энцефалопатиями не всегда соответствовали клинике ЭРиЭЭ [8] – вероятно, разные варианты нуклеотидной последовательности в гене SPTAN1 могут объяснять клиническую вариабельность. Попытки установить генотип-фенотипические корреляции позволили предположить, что место и тип мутации в гене могут играть роль в развитии и тяжести поражения нервной системы. Но из-за небольшого числа пациентов со SPTAN1-ассоциированными энцефалопатиями и относительно большого количества описанных к настоящему времени генных мутаций генотип-фенотипические корреляции до сих пор остаются неопределенными.
В статье впервые в России представлены клинические случаи наблюдения 6 пациентов со SPTAN1-ассоциированными энцефалопатиями.
С 2020 г. 4 пациента с выявленными вариантами нуклеотидной последовательности в гене SPTAN1 наблюдаются в психоневрологическом отделении ГБУЗ «Научно-практический центр специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого» ДЗМ, 1 пациент – в ФГАУ «Национальный медицинский исследовательский центр нейрохирургиимимени академика Н.Н. Бурденко» Минздрава России и 1 пациент – в СПб ГБУЗ «Детский городской многопрофильный клинический специализированный центр высоких медицинских технологий».
Ведение пациентов осуществлялось сообразно принципам Хельсинкской декларации Всемирной медицинской ассоциации (Форталеза, Бразилия, 2013 г.). От родителей пациентов получено информированное согласие на проведение генетического исследования (полноэкзомное секвенирование).
Мальчик 2 лет 6 мес наблюдался с диагнозом: «Синдром детского церебрального паралича, атактическая форма (4-й уровень по шкале GMFCS1). Задержка психоречевого развития. Эпилепсия – вероятно, генетическая мультифокальная, клинико-медикаментозная ремиссия».
Анамнез
Ребенок от второй беременности, протекавшей с угрозой прерывания на 16-й и 22-й неделях гестации. Роды первые в срок, самостоятельные. Масса тела при рождении 3070 г, длина 50 см.
Дебют приступов в 5 мес, когда появились серийные вздрагивания с заведением глазных яблок вверх, в дальнейшем присоединились генерализованные тонико-клонические приступы.
ЭЭГ и МРТ
По данным электроэнцефалографии (ЭЭГ) в 2 года 6 мес регистрировались генерализованные и региональные разряды эпилептиформной активности, локализованные в левой и правой центрально-теменных и иных (реже) областях, независимо, сгруппированного характера, в виде комплексов «пик – волна» (рис. 1).
По данным магнитно-резонансной томографии (МРТ) головного мозга структурных изменений не выявлено (рис. 2).
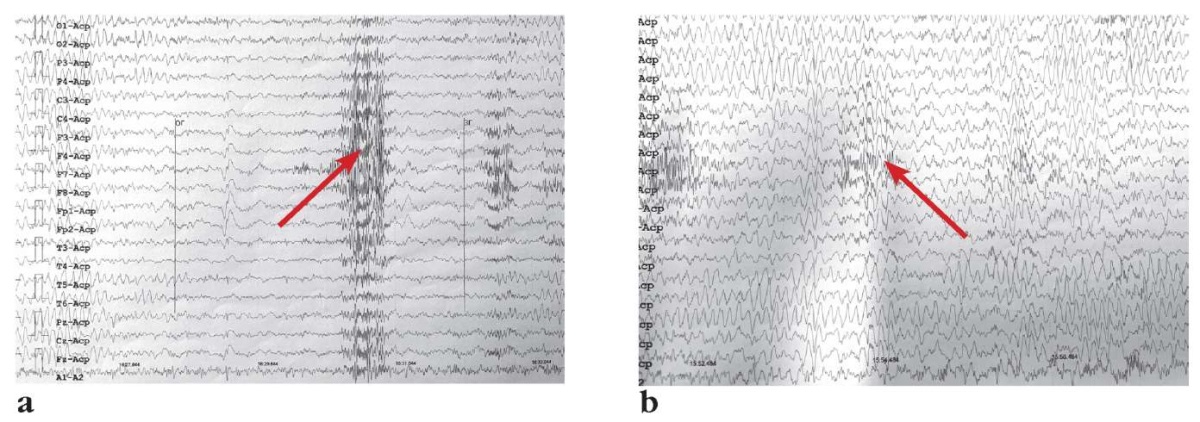
Рисунок 1. Данные электроэнцефалографии
Пациента 1 (2 года 6 мес) на фоне приема вигабатрина:
a – стрелкой указаны генерализованные разряды эпилептиформной активности
в левой и правой центрально-теменных областях;
b – стрелкой указаны региональные разряды эпилептиформной активности
сгруппированного характера в виде комплексов «пик–волна»
Figure 1. Patient 1 (aged 2 years 6 months).
Electroencephalography data upon vigabatrin administration:
a – the arrow indicates generalized discharges of epileptiform activity
in the left and right central parietal regions;
b – the arrow indicates the regional discharges of grouped epileptiform activity
in the form of peak-wave complexes

Рисунок 2. Магнитно-резонансная томограмма головного мозга
Пациента 1 (2 года 6 мес).
Cтруктурные изменения в головном мозге отсутствуют
Figure 2. Patient 1 (aged 2 years 6 months).
Brain magnetic resonance imaging. No brain structural changes
Терапия
Назначена противосудорожная терапия вальпроевой кислотой, проведен курс гормонотерапии (гидрокортизон) в течение 1 мес, на фоне которого отмечалось уменьшение частоты и длительности приступов. В терапию добавлен леветирацетам, приступов не было, а через 1,5 мес они возобновились в прежнем объеме.
На фоне введения в терапию вигабатрина приступы были полностью купированы, в психомоторном развитии отмечена положительная динамика. На ЭЭГ эпиактивность не зарегистрирована. Леветирацетам и топирамат отменены.
Неврологический статус
В неврологическом статусе на момент наблюдения в 2 года 6 мес: альтернирующее расходящееся косоглазие. Осмысленного контакта нет. Речи нет. Указательного жеста нет. Команды не понимает. Моторное развитие: удерживает голову, переворачивается, сидит, стоит, ползает, ходит с поддержкой на широко расставленных ногах. Спонтанная двигательная активность симметричная, хаотичная. Стереотипии. Интерес к окружающему снижен.
В 2 года 6 мес ходит сам. Целенаправленная деятельность бедная. Стереотипии. Контакт с окружающими кратковременный. Речи нет. Мычащие звуки и причмокивания. Манипуляции игрушками примитивные. Жестовый диалог очень скудный. Навыков опрятности и самообслуживания нет. Волосы тонкие. Глазные щели короткие. Прямые длинные ресницы. Альтернирующий страбизм. Взгляд в глаза фиксирует кратковременно. Ушные раковины с утолщенным и диспластичным завитком. Воронкообразная деформация грудной клетки. Гипотония. Тонкие конусовидные пальцы. Клинодактилия мизинцев.
Девочка 9 лет наблюдалась с рождения с диагнозом: «Эпилепсия фокальная с эпилептическими спазмами со статусным, фармакорезистентным течением приступов. Нарушение психо-интеллектуального и речевого развития (сенсомоторная алалия)».
Анамнез
Ребенок от второй беременности в неродственном браке (первая беременность замершая в 7 мес, роды мертвым плодом), протекавшей на фоне угрозы прерывания, анемии 2-й степени, фетоплацентарной недостаточности с 32-й недели гестации, сопровождавшейся внутриутробной задержкой развития плода. Роды вторые самостоятельные, срочные на 41-й неделе гестации. Масса тела при рождении 2140 г, длина 49 см. Оценка по шкале Апгар 7/8 баллов. Развивалась с задержкой психомоторного развития.
Девочка больна с рождения, когда на 2-е сутки жизни возникли тонико-клонические судороги на фоне перинатального поражения центральной нервной системы (ЦНС), синдрома угнетения, геморрагической болезни новорожденных, задержки внутриутробного развития 3-й степени.
Терапия
Получала фенобарбитал. В 2 мес после перенесенной кори присоединились фокальные приступы в виде версии головы и глаз вправо, миоклонических подергиваний в правой кисти длительностью до 3–5 мин. К терапии добавлена вальпроевая кислота в форме сиропа с временным положительным эффектом.
Далее проводилась коррекция противосудорожной терапии, присоединен леветирацетам без выраженного эффекта, после введения в терапию бензодиазепинов приступы купированы. В возрасте 3 лет на фоне фебрильной температуры вновь статус тонико-клонических приступов, купированы в отделении реанимации и интенсивной терапии введением диазепама. Ребенок утратил ранее приобретенные навыки. Диагностирован спастический гемипарез справа.
В дальнейшем судороги со статусным течением повторялись ежегодно. В настоящее время временный положительный эффект достигается комбинацией препаратов леветирацетам, лакосамид, вигабатрин.
ЭЭГ, ТМС и МРТ
По данным ЭЭГ в 9 лет регистрируется региональная эпилептиформная активность в задневисочно-затылочной области слева с распространением на левую теменную область на фоне замедления. Фокальный асимметричный S>D короткий тонический приступ (staring – гипомоторный приступ, легкое тоническое напряжение левой руки длительностью 1,5 мин) (рис. 3).
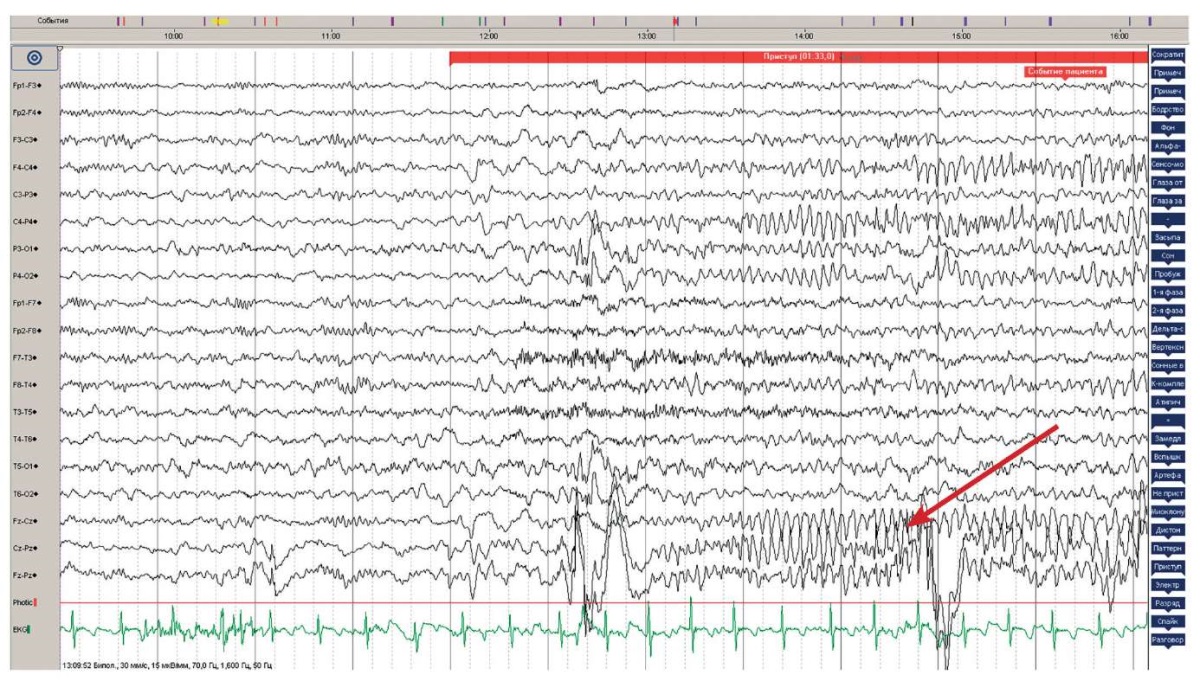
Рисунок 3. Данные электроэнцефалографии Пациентки 2 (9 лет).
Стрелкой указана эпилептиформная активность
в задневисочно-затылочной области слева
с распространением на левую теменную область на фоне замедления
Figure 3. Patient 2 (aged 9 years). Electroencephalography data.
The arrow indicates epileptiform activity in the left posterior occipital region
with spread to the left parietal region during deceleration
Транскраниальная магнитная стимуляция (ТМС) в 9 лет: данных за наследственные аминоацидопатии, органические ацидурии, дефекты митохондриального бета-окисления не выявлено.
МРТ головного мозга (3 Тл) в 9 лет: резидуальные глиозно-атрофические изменения в теменно-затылочных областях больших полушарий головного мозга (рис. 4).
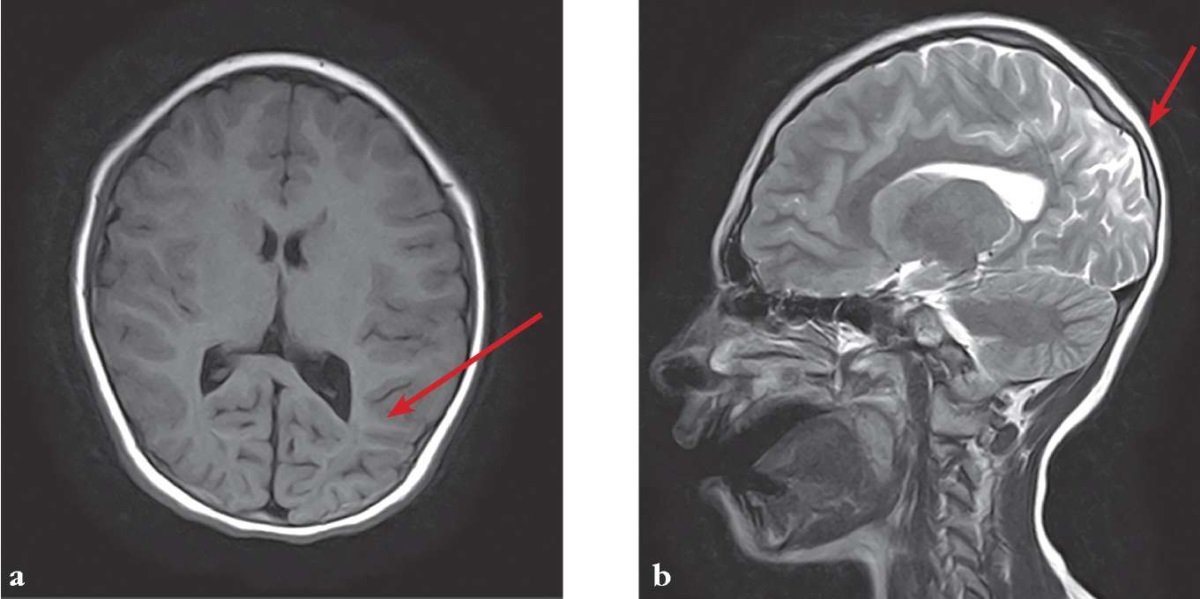
Рисунок 4. Данные магнитно-резонансной томографии Пациентки 2 (9 лет).
Стрелками указаны глиозно-атрофические изменения
в теменно-затылочных областях больших полушарий головного мозга (a, b)
Figure 4. Patient 2 (aged 9 years).
Magnetic resonance imaging data. Arrows indicate glial-atrophic changes
in the parieto-occipital regions of the cerebral hemispheres (a, b)
Неврологический статус
В неврологическом статусе задержка психомоторного развития с рождения. В 11 лет микрокрания. Густые волосы, низкий рост волос на лбу. Диспластичные ушные раковины с утолщенным завитком. Гиперметропический астигматизм (пользуется очками). Короткий фильтр. Аномалия прикуса. Узкие плечи. Тонкие конусовидные пальцы. Вальгусная установка стоп. Дифференцировка на «свои/чужие» сохранена. Речи нет. Жестовый диалог примитивный. Мелкая моторика не развита, движения неловкие. Тремор конечностей. Эмоциональна. Навязчива. Понимает ситуативную бытовую инструкцию. Навыки опрятности частично сформированы. Походка дискоординированная на широко расставленных ногах с вальгусной установкой стоп.
Мальчик 13 лет наблюдался с диагнозом: «Эпилепсия неизвестная, комбинированная (с признаками фокальной и генерализованной), фармакорезистентное течение. Эпилептическая энцефалопатия. Расстройство вегетативной нервной системы».
Анамнез
Ребенок от третьей беременности, протекавшей без особенностей. Роды третьи в срок, самостоятельные. Масса тела при рождении 3200 г, длина 51 см. Психомоторное развитие по возрасту (со слов родителей, поскольку ребенок не говорит по-русски).
Дебют приступов в 2 года, установлен диагноз «посттравматическая фокальная эпилепсия, когнитивные нарушения». Получал противоэпилептические препараты (ПЭП) без эффекта. При первой госпитализации в психоневрологическое отделение на фоне приема сультиама – выраженное улучшение. Данный препарат не зарегистрирован в Российской Федерации, в связи с чем была проведена врачебная комиссия, на которой определена необходимость приема сультиама.
МРТ и ЭЭГ
По данным МРТ головного мозга в 13 лет структурных изменений не выявлено (рис. 5).
ЭЭГ в 13 лет: регистрируется эпилептиформная активность в левых лобных отделах с распространением на соседние отделы, тенденцией к вторичной билатеральной синхронизации, в правых центральных отделах – с распространением на вертексные в левых теменно-задневисочных отделах. Конфигурация разрядов напоминает «доброкачественные эпилептиформные разряды детского возраста». Индекс представленности в бодрствовании и во сне средний (30–50%) (рис. 6).

Рисунок 5. Данные магнитно-резонансной томографии Пациента 3 (13 лет).
Структурные изменения в головном мозге отсутствуют (a, b)
Figure 5. Patient 3 (aged 13 years). Magnetic resonance imaging data.
No brain structural changes observed (a, b)
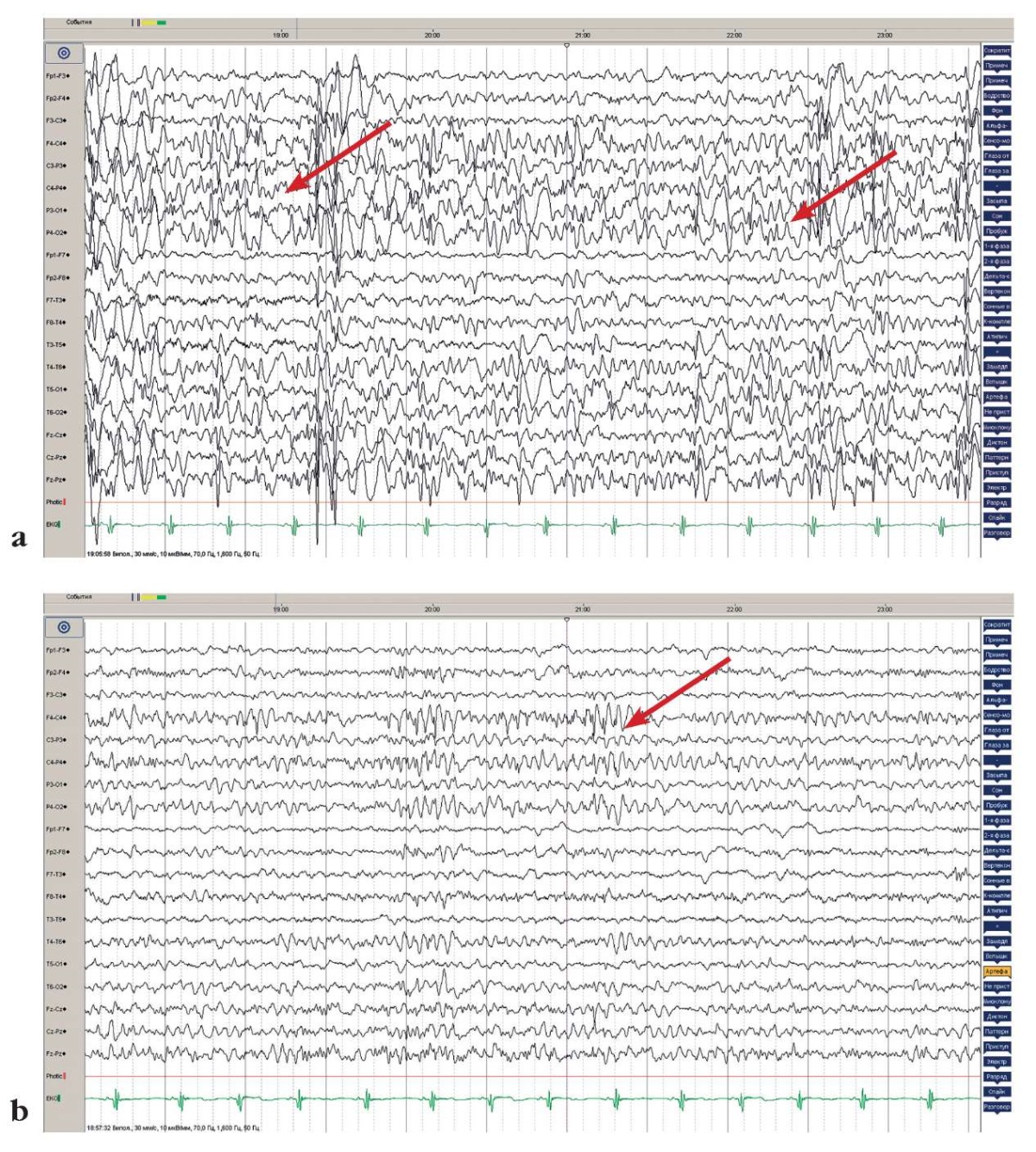
Рисунок 6. Данные электроэнцефалографии Пациента 3 (13 лет):
a – стрелками указана эпилептиформная активность
в левых лобных отделах с распространением на соседние отделы
и тенденцией к вторичной билатеральной синхронизации,
в правых центральных отделах – с распространением на вертексные
в левых теменно-задневисочных отделах;
b – стрелкой указаны «доброкачественные эпилептиформные разряды
детского возраста»,
индекс представленности в бодрствовании и во сне средний (30–50%)
Figure 6. Patient 3 (aged 13 years). Electroencephalography data:
a – arrows indicate epileptiform activity in the left frontal regions
with spread to neighboring regions tended to secondary bilateral synchronization,
in the right central regions spreading to the vertex
in the left parieto-posterior temporal regions;
b – the arrow indicates “benign epileptiform discharges of childhood”,
with average sleep-wake representation index (30–50%)
Неврологический статус
В неврологическом статусе без особенностей. Сохраняется когнитивный дефицит.
Мальчик 8 лет 3 мес наблюдался с диагнозом: «Эпилепсия криптогенная генерализованная, фармакорезистентное течение. Эпилептическая энцефалопатия. Нарушение психоинтеллектуального развития».
Анамнез
Ребенок от второй беременности, протекавшей без особенностей. Роды первые в срок, самостоятельные. Масса тела при рождении 3360 г, длина 53 см. По шкале Апгар 7/8 баллов. Психомоторное развитие до 4 лет по возрасту.
Дебют заболевания в 4 года на фоне полного здоровья, когда при засыпании развился приступ: миоклонические подергивания левой половины лица с переходом в тонико-клонический приступ длительностью до 1 мин. Назначена вальпроевая кислота, приступов не было до 5 лет, затем они возобновились.
Терапия
Фармакологический анамнез: вальпроевая кислота – приступов не было около 1 года, леветирацетам – без эффекта, окскарбазепин – аггравация, топирамат – без эффекта.
ЭЭГ и МРТ
По данным ЭЭГ в 8 лет регистрируется эпилептиформная активность в правой лобно-височной области, периодически с миграцией акцента на теменно-центральные отделы, в левых височно-центральных отделах в виде диффузных разрядов с правосторонним акцентом. Во сне эпилептиформная активность представлена диффузными (генерализованными) разрядами как одиночного, так и сгруппированного, ритмичного характера, на некоторых эпохах занимает до 90% записи.
По данным МРТ головного мозга определены зоны дисгармоничного формирования кортекса в полюсе левой лобной доли, единичные очаговые изменения вещества головного мозга, явления перивентрикулярного глиоза боковых желудочков – вероятно, резидуального характера.
Неврологический статус
В неврологическом статусе очаговых нарушений нет. Когнитивный дефицит. Гипердинамический синдром.
Девочка 5 лет 11 мес наблюдалась с диагнозом: «Эпилепсия симптоматическая, фокальные приступы с генерализацией».
Анамнез
Ребенок от второй беременности, протекавшей с угрозой прерывания на 12-й неделе. Роды вторые, самостоятельные. Масса тела при рождении 2990 г, длина 50 см. По шкале Апгар 8/8 баллов. На 1-е сутки отмечалось внутрижелудочковое кровоизлияние 2-й степени.
Дебют приступов в возрасте 1 мес в виде «таращенья» глаз, причмокиваний, гиперсаливации с тоническим напряжением и отведением в стороны правых конечностей длительностью несколько секунд.
Терапия
Фармакоанамнез: фенобарбитал – ремиссия 14 дней, вальпроевая кислота – без эффекта, леветирацетам – ремиссия 3 нед, зонегран – побочное явление в виде задержки мочи, вигабатрин – без эффекта, клобазам – без эффекта.
В 2,5 года девочке введен метилпреднизолон с положительной динамикой в виде сокращения приступов. Однако на фоне снижения дозы гормона стали отмечаться статусные приступы. Также предпринята попытка введения пациентке кетогенной диеты, но на 2-е сутки у ребенка отмечалась острая задержка мочи. Диета отменена, состояние нормализовалось в течение 1 сут. Затем появились фокальные приступы в виде заведения глаз с ороалиментарными автоматизмами и вегетативным компонентом (покраснение вокруг глаз). В настоящее время у ребенка наблюдаются фокальные моторные приступы в виде клоний в правой руке с отведением правого угла рта длительностью до 6 мин.
ЭЭГ и МРТ
По данным ЭЭГ в 5 лет в бодрствовании зарегистрирован основной ритм частотой 7–8 Гц. Также отмечается эпилептиформная активность на фоне продолженного замедления в левой лобно-центральной области с диффузным распространением. Во сне картина модифицированной гипсаритмии, зонально-акцентуированный вариант (лобные отделы полушарий) (рис. 7).
По данным МРТ в 5 лет выявлены признаки гипоксически-ишемического поражения ЦНС, смешанная гидроцефалия (рис. 8).
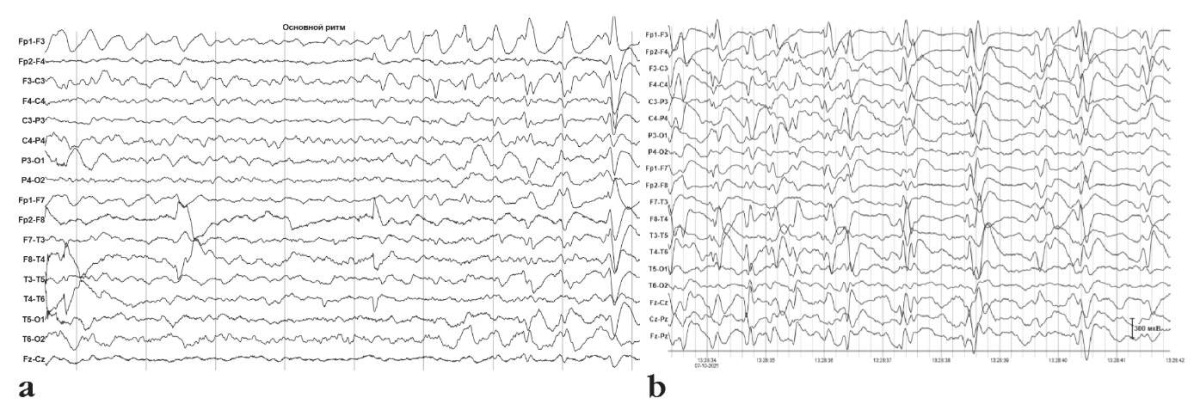
Рисунок 7. Данные электроэнцефалографии Пациентки 5 (5 лет 11 мес):
a – эпилептиформная активность на фоне продолженного замедления
в левой лобно-центральной области с диффузным распространением;
b – картина модифицированной гипсаритмии,
зонально-акцентуированный вариант (лобные отделы полушарий) во сне
Figure 7. Patient 5 (aged 5 years 11 months). Electroencephalography data:
a – epileptiform activity during continued deceleration
in the left fronto-central region with diffuse spread;
b – a modified hypsarrhythmia, sleep zonal accentuated variant
(frontal parts of the hemispheres)

Рисунок 8. Данные магнитно-резонансной томографии Пациентки 5 (5 лет 11 мес).
Стрелкой указаны признаки гипоксически-ишемического поражения
центральной нервной системы, смешанная гидроцефалия
Figure 8. Patient 5 (aged 5 years 11 months).
Magnetic resonance imaging data.
The arrow indicates signs of hypoxic-ischemic damage
to the central nervous system, mixed hydrocephalus
Неврологический статус
В неврологическом статусе микроцефалия (46,5 см). Периодическое сходящееся косоглазие, мышечная гипотония. Грубая задержка психомоторного развития: ребенок удерживает голову в положении сидя и в положении на животе, при тракции за руки не группируется, не ползает, не ходит. Девочка следит за предметами (фонарик, игрушка) и берет их в руки, может переложить. Активной речи нет, но есть единичные слоги.
Девочка 4 лет 2 мес наблюдается с 3 мес по поводу инфантильных спазмов.
Анамнез
Беременность вторая в неродственном браке. Протекала без особенностей. Оперативные плановые роды в срок (тазовое предлежание), масса тела 3100 г, длина 51 см. По шкале Апгар 8/9 баллов.
До момента начала приступов в течение 2 мес была беспокойной. Психомоторное развитие по возрасту. Получала курс тетракозактида в дозе 0,3 мл внутримышечно 1 раз в неделю на 6 мес (с положительной динамикой). Данный препарат не зарегистрирован в Российской Федерации, в связи с чем была проведена врачебная комиссия, на которой определена необходимость приема тетракозактида.
ТМС и ЭЭГ
Селективный метаболический скрининг методом ТМС в 2 года – норма. Рецидив приступов в возрасте 3 лет после сна с фиксацией взора, адверсией глазных яблок. По данным ЭЭГ в бодрствовании и с нарастанием во сне зарегистрирована эпилептическая активность: комплексы «острая – медленная волна» в левой височно-лобной области с последующим диффузным распространением. Психомоторное развитие по возрасту. Очаговой неврологической симптоматики нет.
ЭЭГ в 4 года: субдоминирует бета-активность с индексом до 20%, представленная диффузно, с акцентом по передним отделам. Эпиактивность не зарегистрирована. Приступы миоклонические фокальные в левой половине лица с фиксацией взора длительностью до 2 мин, «замирания».
Терапия
Назначен вигабатрин в дозе 250 мг 2 раза в день на 3 мес с последующим снижением дозы и присоединением ламотриджина до 4,5 мг/кг в 2 приема. Через 1 мес рецидив приступов до 10–12 в сутки. По данным ЭЭГ в левой височной области регистрируется регионарная эпилептиформная активность, периодически кластерного характера. В терапию введена вальпроевая кислота 30 мг/кг, увеличена доза ламотриджина до 7 мг/кг. На представленных МРТ-изображениях в 4 года – фокальная кортикальная дисплазия левой височной доли.
Учитывая сохраняющиеся приступы, начаты постепенная отмена вальпроевой кислоты и введение в терапию окскарбазепина в дозе до 600 мг/сут.
Неврологический статус
Неврологический статус в возрасте 4 лет: внешне и физически ребенок развит (ходит, бегает, прыгает и т.д.). В речи знает много отдельных слов, но только недавно начала говорить короткими фразами. На данный момент приступы купированы, девочка принимает вигабатрин и окскарбазепин.
Принимая во внимание раннее возникновение судорог, неспецифических фенотипических проявлений заболевания, все пациенты консультированы врачом-генетиком, при информированном согласии родителей рекомендовано проведение полноэкзомного секвенирования. На основании проведенного анализа у наблюдаемых нами детей выявлены варианты нуклеотидной последовательности в гене SPTAN1.
Клинические и молекулярные характеристики пациентов представлены в таблице 1.
Таблица 1. Клинические и молекулярные характеристики
пациентов с вариантами нуклеотидной последовательности в гене SPTAN1
Table 1. Clinical and molecular characteristics
of patients with SPTAN1 gene nucleotide sequence variants
|
Параметр / Parameter |
Пациент / Patient |
|||||
|
1 |
2 |
3 |
4 |
5 |
6 |
|
|
Пол / Gender |
Мужской / Male |
Женский / Female |
Мужской / Male |
Мужской / Male |
Женский / Female |
Женский / Female |
|
Возраст / Age |
2 года 6 мес / |
11 лет / 11 years |
13 лет / 13 years |
8 лет 3 мес / 8 years 3 months |
5 лет 11 мес / |
4 года 2 мес / |
|
Вариант нуклеотидной последовательности (GRCh37/hg19) / Nucleotide |
c.1595A>G (Lys532Arg) гетерозигота |
c.4027C>T (Arg1343Cys) гетерозигота |
c.1375C>T (Arg459Cys) гетерозигота |
с.6577-1G>C гетерозигота |
c.6893_6901del гетерозигота |
c.1362G>C (Glu454Asp) гетерозигота |
|
Возраст начала приступов / |
5 мес / 5 months |
2-е сутки жизни / 2 days of life |
2 года / 2 years |
4 года / 4 years |
1 мес / 1 month |
3 мес / 3 months |
|
Тип судорог / |
Генерализованные тонико-клонические / Generalized tonic-clonic |
Тонико-клонические, миоклонические, фокальные, гипомоторные / |
Тонико-клонические, приступы в виде сильной головной боли / |
Миоклонические подергивания, приступы по типу негативного миоклонуса (атонический) с миоклониями век, тонико-клонические приступы / Myoclonic twitches, negative myoclonus (atonic) type seizures with eyelid myoclonus, tonic-clonic seizures |
Фокальные моторные с автоматизмами и вегетативным компонентом, фокальные моторные / Focal motor with automatisms and vegetative component, focal motor |
Инфантильные спазмы, миоклонические фокальные левой половины лица / Infantile spasms, left face-half focal myoclonic |
|
ЭЭГ / EEG |
Норма / Normal |
Резидуальные глиозно-атрофические изменения в теменно-затылочных областях больших полушарий / Residual gliosis-atrophic changes in parieto-occipital regions of the cerebral hemispheres |
Норма / Normal |
Картина зоны дисгармоничного формирования кортекса в полюсе левой лобной доли, единичные очаговые изменения вещества головного мозга, перивентрикулярный глиоз боковых желудочков / Pattern of zonal aberrant cortical development in the pole of the left frontal lobe, single focal changes in brain substance, periventricular gliosis of the lateral ventricles |
Признаки гипоксически-ишемического поражения ЦНС, смешанная гидроцефалия / Signs of hypoxic-ischemic CNS damage, mixed hydrocephalus |
ФКД левой височной доли / FCD of the left temporal lobe |
|
Моторное развитие / |
Задержка моторного развития, мышечная гипотония / Delayed motor development, muscle hypotension |
Задержка моторного развития, мышечная гипотония / Delayed motor development, muscle hypotension |
Моторное развитие по возрасту / Age-related motor development |
До 4 лет по возрасту, затем с выраженной задержкой / By age 4 years old: age-related development followed by profound retardation |
Грубая задержка психомоторного развития, мышечная гипотония / Gross psychomotor retardation, muscular hypotension |
Моторное развитие по возрасту / Age-related motor development |
|
Психоречевое развитие / |
Задержка психоречевого развития / Delayed psycho-speech development |
Задержка психоречевого развития, речи нет / Delayed psycho-speech development, no speech |
Задержка психоречевого развития / Delayed psycho-speech development |
До 4 лет по возрасту, затем с выраженной задержкой / By age 4 years old: age-related development followed by profound retardation |
Выраженная задержка психоречевого развития / Severe delayed psycho-speech development |
Задержка психоречевого развития / Delayed |
|
Эффективность терапии / Therapeutic efficiency |
Вальпроевая кислота – б/э, |
Леветирацетам, лакосамид, вигабатрин – с временным эффектом / Levetiracetam, lacosamide, vigabatrin – temporary effect |
Ламиктал® – б/э, |
Леветирацетам – б/э, трилептал – аггравация, топирамат – б/э // Levetiracetam – NE, trileptal – aggravation, topiramate – NE |
Вальпроевая кислота – б/э, леветирацетам – 3 нед ремиссия, вигабатрин – б/э, клобазам – б/э // Valproic acid – NE, levetiracetam – 3 week-remission, vigabatrin – NE, clobazam – NE |
Вигабатрин и окскарбазепин – с положительным эффектом / Vigabatrin and oxcarbamazepine – positive effect |
Примечание. ЭЭГ – электроэнцефалография;
МРТ – магнитно-резонансная томография;
ЦНС – центральная нервная система;
ФКД – фокальная кортикальная дисплазия;
б/э – без эффекта.
Note. EEG – electroencephalography;
MRI – magnetic resonance imaging;
CNS – central nervous system;
FCD – focal cortical dysplasia;
NE – no effect.
То, что в основе развития эпилепсии, возможно, лежит повреждение генома, предполагалось еще до периода активного развития и внедрения технологий генетического тестирования [9]. В настоящее время уже известно, что с развитием эпилепсии ассоциировано более 1000 генов (так называемые моногенные формы, когда мутация в одном гене приводит к заболеванию, и полигенные формы – вовлеченность в патологический процесс нескольких генов) [9].
Следует отметить, что десятилетие назад врачи сталкивались с большими трудностями в отношении генетической диагностики эпилепсии. Это связано с тем, что, во-первых, значительная часть генов еще не была идентифицирована, а во-вторых, те гены, структура которых была установлена, имеют больший размер, а это, в свою очередь, обусловливает существенные экономические затраты на секвенирование их по отдельности. Спустя годы удалось добиться повышения эффективности и снижения экономических затрат благодаря развитию и внедрению в клиническую практику массового параллельного секвенирования, или секвенирования следующего поколения (англ. next generation sequencing, NGS). Ранняя генетическая диагностика не только может сократить время и общие затраты на постановку диагноза, но и на основе полученной информации позволит определять прогноз заболевания и выбор таргетной терапии.
Ген αII-спектрина, SPTAN1 (OMIM: 182810), кодирует мембранный каркасный белок, играющий важную роль в поддержании целостности миелинизированных аксонов, развитии аксонов и синаптогенезе. Патогенные de novo варианты в гене спектрина αII (SPTAN1) ответственны за широкий спектр нарушений развития нервной системы, включая эпилептическую энцефалопатию с ранним началом и прогрессирующей атрофией головного мозга, тяжелый интеллектуальный дефицит с пороками развития мозжечка и относительно более мягкий фенотип с эпилепсией или без нее [8][10].
Большинство случаев с вариантами в гене SPTAN1 ассоциированы с ЭРиЭЭ, однако в последние годы на основании наблюдения пациентов их выявление связывают также с развитием наследственной моторной невропатии и аутосомно-рецессивной наследственной спастической параплегией [11–13]. У больных с эпилептической энцефалопатией патогенные варианты в гене SPTAN1 приводят к нарушению развития головного мозга и белого вещества, что в конечном итоге приводит к диффузной атрофии коры, мозолистого тела, ствола мозга и мозжечка посредством доминантно-негативного механизма, вызывающего агрегацию комплексов спектрина [14].
S. Syrbe et al. [8] проанализировали данные 20 пациентов с патогенными или вероятно патогенными вариантами в гене SPTAN1 и сравнили их с вариантами в ранее опубликованных исследованиях. Авторы показали, что 62% больных детей имеют ЭРиЭЭ с ранним началом рецидивирующих фармакорезистентных судорог, тяжелую задержку развития. У нескольких пациентов был описан синдром Веста с типичными ЭЭГ-проявлениями, которые имели тенденцию развиваться в неорганизованную медленную фоновую активность с частыми мультифокальными спайками. Замедленная и неполная миелинизация, связанная с атрофией головного мозга, была основным проявлением заболевания, обнаруживаемым на МРТ, и может считаться отличительной чертой SPTAN1-ассоциированной энцефалопатии. Смерть в первые годы жизни часто отмечалась у данных пациентов в исследовании. Однако примерно в 30% случаев фенотип был легким, с судорогами, начавшимися после младенчества, с относительно хорошим ответом на ПЭП и отсутствием риска ранней смерти [8].
Интересно, что варианты в гене SPTAN1, выявленные у пациентов с легким течением заболевания, были локализованы далеко от С-терминального региона. Напротив, в большинстве тяжелых случаев обнаружены мутации с делецией/дупликацией внутри рамки считывания, расположенные в последних двух повторах спектрина αII в С-терминальном регионе (рис. 9). Поскольку эти повторы необходимы для α/β-связи гетеродимера спектрина, считается, что образование измененного гетеродимера между спектринами вызывает нестабильность и агрегацию спектриновых каркасов [15]. У таких больных эпилепсия является важным клиническим признаком и проявляется инфантильными спазмами, фармакорезистентностью к ПЭП или поддающимися лечению генерализованными (миоклоническими, тоническими, атоническими) и фокальными приступами с нарушением сознания [8]. На ЭЭГ выявляют гипсаритмию, генерализованное замедление фона и мультифокальные эпилептиформные аномалии. Однако недавно V. Gartner et al. сообщили о 2 пациентах, несущих мутации в критических повторах спектрина для развития эпилепсии без истории приступов [10].
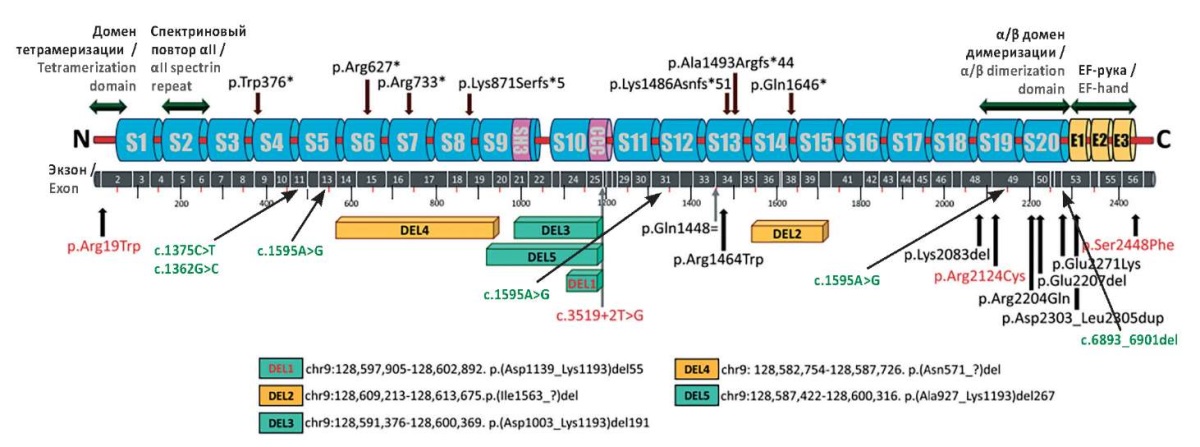
Рисунок 9. Ранее описанные варианты нуклеотидной последовательности
в гене SPTAN1 и доменная структура кодируемого αII-спектрина (адаптровано по [11]).
Зеленым цветом выделены варианты нуклеотидной последовательности,
выявленные у наблюдаемых нами пациентов
Figure 9. Previously described variants of SPTAN1 gene nucleotide sequence
and αII-spectrin-encoded domain structure (adapted from [11]).
Variants of the nucleotide sequence identified
in the patients we observed are highlighted in green
Исследования in vitro, проведенные как на трансфицированных первичных нейронах, так и на лимфобластоидных клетках пациента с мутациями в двух последних повторах спектрина αII, показали, что гетеродимеры спектрина αII/βII, содержащие мутантный спектрин, более нестабильны, чем гетеродимеры с нормальным спектрином. Кроме того, нестабильность была связана с образованием агрегатов [8]. Это приводит к потере целостности начального сегмента аксона, который необходим для нормального развития и функционирования нервной системы, и не удивительно, что его повреждение может вызвать тяжелое течение ЭРиЭЭ [16].
Мы наблюдали 6 пациентов с ранним началом судорог (от 2 сут жизни до 4 лет), задержкой психомоторного и речевого развития. При проведении полноэкзомного секвенирования были идентифицированы варианты нуклеотидной последовательности в гене SPTAN1.
Анализ клинического течения заболевания подтвердил полученные ранее данные о связи вариантов в гене с поражением ЦНС. Кроме того, на российской когорте пациентов установлена связь между локализацией варианта и течением заболевания. Так, в трех наблюдениях (пациенты 1, 3, 6) варианты в гене локализованы в районе N-конца, они характеризуются более легким течением заболевания, отсутствием структурных изменений головного мозга (пациенты 1 и 3), мягким когнитивным дефицитом и положительным эффектом в отношении приступов при применении ПЭП. В случаях вариантов вблизи C-терминального региона (пациенты 2, 4, 5) тяжесть течения заболевания обусловлена статусным течением приступов (пациент 2), выраженной задержкой психомоторного и речевого развития, изменениями на МРТ головного мозга (глиозно-атрофические изменения – пациент 2, дисгормоничное формирование кортекса, единичные очаговые изменения вещества головного мозга, явления перивентрикулярного глиоза боковых желудочков – пациент 4) и фармакорезистентностью.
Впервые в России представлены клинические случаи SPTAN1-ассоциированной энцефалопатии. Молекулярная диагностика у пациентов с нарушениями развития нервной системы часто вызывает затруднения как из-за неспецифического фенотипа, так и вследствие генетической гетерогенности. Редкие варианты нуклеотидной последовательности в генах в такой группе заболеваний удается в большинстве случаев обнаружить при направлении больных на полноэкзомное секвенирование. Нам удалось расширить фенотипический и генетический спектр вариантов нуклеотидной последовательности в гене SPTAN1.
Таким образом, описанные нами клинические случаи показывают, что варианты в гене, кодирующем αII спектрин, ассоциированы с фенотипом энцефалопатии развития и эпилептической энцефалопатии. Клинический анамнез, данные ЭЭГ и МРТ пациентов схожи с ранее описанными в научной литературе. Наши наблюдения подтверждают, что варианты в гене SPTAN1, локализованные ближе к С-терминальному региону, ассоциированы с более тяжелым фенотипом, а варианты вблизи N-региона – с более легким течением заболевания без структурных аномалий головного мозга. Однако в будущем необходимы дальнейшие исследования для лучшего понимания генотип-фенотипических корреляций при SPTAN1-ассоциированной энцефалопатии.
1. GMFCS (англ. Gross Motor Function Classification System) – система классификации общих двигательных функций.
1. Berg A.T., Berkovic S.F., Brodie M.J., et al. Revised terminology and concepts for organization of seizures and epilepsies: Report of the ILAE Commission on Classification and Terminology, 2005– 2009. Epilepsia. 2010; 51: 676–85. https://doi.org/10.1111/j.15281167.2010.02522.x.
2. Radaelli G., de Souza Santos F., Borelli W.V., et al. Causes of mortality in early infantile epileptic encephalopathy: a systematic review. Epilepsy Behav. 2018; 85: 32–6. https://doi.org/10.1016/j.yebeh.2018.05.015.
3. Pavone P., Corsello G., Ruggieri M., et al. Benign and severe early-life seizures: a round in the first year of life. Ital J Pediatr. 2018; 44 (1): 54. https://doi.org/10.1186/s13052-018-0491-z.
4. Nieh S.E., Sherr E.H. Epileptic encephalopathies: new genes and new pathways. Neurotherapeutics. 2014; 11 (4): 796–806. https://doi.org/10.1007/s13311-014-0301-2.
5. Zhang R., Zhang C., Zhao Q., Li D. Spectrin: structure, function and disease. Sci China Life Sci. 2013; 56 (12): 1076–85. https://doi.org/10.1007/s11427-013-4575-0.
6. Wang Y., Ji T., Nelson A.D., et al. Critical roles of αII spectrin in brain development and epileptic encephalopathy. J Clin Invest. 2018; 128 (2): 760–73. https://doi.org/10.1172/JCI95743.
7. Begg G.E., Harper S.L., Morris M.B., Speicher D.W. Initiation of spectrin dimerization involves complementary electrostatic interactions between paired triple-helical bundles. J Biol Chem. 2000; 275 (5): 3279–87. https://doi.org/10.1074/jbc.275.5.3279.
8. Syrbe S., Harms F.L., Parrini E., et al. Delineating SPTAN1 associated phenotypes: from isolated epilepsy to encephalopathy with progressive brain atrophy. Brain. 2017; 140 (9): 2322–36. https://doi.org/10.1093/brain/awx195.
9. Møller R.S., Dahl H.A., Helbig I. The contribution of next generation sequencing to epilepsy genetics. Expert Rev Mol Diagn. 2015; 15 (12): 1531–8. https://doi.org/10.1586/14737159.2015.1113132.
10. Gartner V., Markello T.C., Macnamara E., et al. Novel variants in SPTAN1 without epilepsy: an expansion of the phenotype. Am J Med Genet A. 2018; 176 (12): 2768–76. https://doi.org/10.1002/ajmg.a.40628.
11. Morsy H., Benkirane M., Cali E., et al. Expanding SPTAN1 monoallelic variant associated disorders: from epileptic encephalopathy to pure spastic paraplegia and ataxia. Genet Med. 2023; 25 (1): 76–89. https://doi.org/10.1016/j.gim.2022.09.013.
12. Beijer D., Deconinck T., De Bleecker J.L., et al. Nonsense mutations in alpha-II spectrin in three families with juvenile onset hereditary motor neuropathy. Brain. 2019; 142 (9): 2605–16. https://doi.org/10.1093/brain/awz216.
13. Leveille E., Estiar M.A., Krohn L., et al. SPTAN1 variants as a potential cause for autosomal recessive hereditary spastic paraplegia. J Hum Genet. 2019; 64 (11): 1145–51. https://doi.org/10.1038/s10038-0190669-2.
14. Wang Y., Ji T., Nelson A.D., et al. Critical roles of αII spectrin in brain development and epileptic encephalopathy. J Clin Invest. 2018; 128 (2): 760–73. https://doi.org/10.1172/JCI95743.
15. Bennett V., Baines A.J. Spectrin and ankyrin-based pathways: metazoan inventions for integrating cells into tissues. Physiol Rev. 2001; 81 (3): 1353–92. https://doi.org/10.1152/physrev.2001.81.3.1353.
16. Leterrier C. The axon initial segment: an updated viewpoint. J Neurosci. 2018; 38 (9): 2135–45. https://doi.org/10.1523/JNEUROSCI.1922-17.2018.
Кожанова Татьяна Викторовна – к.м.н., доцент кафедры неврологии, нейрохирургии и медицинской генетики им. академика Л.О. Бадаляна педиатрического факультета; ведущий научный сотрудник генетической группы научного отдела, врач – лабораторный генетик
ул. Островитянова, д. 1, Москва 117997
ул. Авиаторов, д. 38, Москва 119620
Жилина Светлана Сергеевна – к.м.н., доцент кафедры неврологии, нейрохирургии и медицинской генетики им. академика Л.О. Бадаляна педиатрического факультета; ведущий научный сотрудник генетической группы научного отдела, врач-генетик
ул. Островитянова, д. 1, Москва 117997
ул. Авиаторов, д. 38, Москва 119620
Мещерякова Татьяна Ивановна – к.м.н., ведущий научный сотрудник генетической группы научного отдела, врач-генетик
ул. Авиаторов, д. 38, Москва 119620
Лукьянова Екатерина Геннадьевна – врач-невролог
ул. Авиаторов, д. 38, Москва 119620
Большакова Екатерина Сергеевна – врач-невролог
ул. Авиаторов, д. 38, Москва 119620
Айвазян Сергей Оганесович – к.м.н., ведущий научный сотрудник генетической группы научного отдела
Scopus Author ID: 35773251400
ул. Авиаторов, д. 38, Москва 119620
Осипова Карина Вартановна – к.м.н., заведующая психоневрологическим отделением
ул. Авиаторов, д. 38, Москва 119620
Власов Павел Александрович – врач-невролог
ул. 4-я Тверская-Ямская, д. 16, Москва 125047
Крапивкин Алексей Игоревич – д.м.н., директор
ул. Авиаторов, д. 38, Москва 119620
Заваденко Николай Николаевич – д.м.н., профессор, заведующий кафедрой неврологии, нейрохирургии и медицинской генетики им. академика Л.О. Бадаляна педиатрического факультета
ул. Островитянова, д. 1, Москва 117997
Кожанова Т.В., Жилина С.С., Мещерякова Т.И., Лукьянова Е.Г., Большакова Е.С., Айвазян С.О., Осипова К.В., Власов П.А., Крапивкин А.И., Заваденко Н.Н. SPTAN1-ассоциированная энцефалопатия развития и эпилептическая энцефалопатия. Эпилепсия и пароксизмальные состояния. 2023;15(3):246–259. https://doi.org/10.17749/2077-8333/epi.par.con.2023.150
Kozhanova T.V., Zhilina S.S., Meshcheryakova T.I., Lukyanova E.G., Bolshakova E.S., Ayvazyan S.O., Osipova K.V., Vlasov P.A., Krapivkin A.I., Zavadenko N.N. SPTAN1-associated developmental and epileptic encephalopathy. Epilepsy and paroxysmal conditions. 2023;15(3):246–259. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2023.150

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































