


Содержание
Перейти к:
 Т. В. Кожанова,
С. С. Жилина,
Л. М. Сушко,
Е. Г. Лукьянова,
К. В. Осипова,
А. И. Крапивкин,
Н. Н. Заваденко
Т. В. Кожанова,
С. С. Жилина,
Л. М. Сушко,
Е. Г. Лукьянова,
К. В. Осипова,
А. И. Крапивкин,
Н. Н. Заваденко https://doi.org/10.17749/2077-8333/epi.par.con.2023.159
Перейти к:
Фокальная эпилепсия является наиболее распространенным типом эпилепсии, на долю которой приходится 60–70% всех случаев данной патологии. В статье представлены два семейных случая фокальной эпилепсии, ассоциированной с вариантами нуклеотидной последовательности в гене DEPDC5. Проведено клинико-генеалогическое обследование, использованы инструментальные (магнитно-резонансная томография, видеоэлектроэнцефалографический мониторинг) и генетический методы исследования. Выявлены варианты нуклеотидной последовательности в гене DEPDC5 у двух пробандов и их отцов с эпилепсией. Фокальная кортикальная дисплазия определена у отца пробанда 1 и у пробанда 2 с резистентным течением эпилепсии и выраженным когнитивным дефицитом. Данные клинические случаи подтверждают, что патогенные варианты в гене DEPDC5 ассоциированы с семейной фокальной эпилепсией, клиническое проявление которой может зависеть от типа выявленной мутации. Изучение генотип-фенотипических корреляций необходимо для назначения терапии. При принятии решения о хирургическом лечении эпилепсии следует проводить генетическое тестирование методом полноэкзомного или полногеномного секвенирования.
Кожанова Т.В., Жилина С.С., Сушко Л.М., Лукьянова Е.Г., Осипова К.В., Крапивкин А.И., Заваденко Н.Н. DEPDC5-ассоциированная семейная фокальная эпилепсия. Эпилепсия и пароксизмальные состояния. 2023;15(4):339-347. https://doi.org/10.17749/2077-8333/epi.par.con.2023.159
Kozhanova T.V., Zhilina S.S., Sushko L.M., Lukyanova E.G., Osipova K.V., Krapivkin A.I., Zavadenko N.N. DEPDC5-related familial focal epilepsy. Epilepsy and paroxysmal conditions. 2023;15(4):339-347. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2023.159
Фокальная эпилепсия является наиболее распространенным типом эпилепсии, на долю которой приходится 60–70% всех случаев данной патологии. Однако этиология более чем половины фокальных эпилепсий остается неясной.
Эпилепсия, связанная с патогенными вариантами нуклеотидной последовательности в гене DEPDC5, представляет собой недавно описанную преимущественно фокальную эпилепсию, связанную с усилением функционирования mTORC1-пути. Варианты в гене DEPDC5 были идентифицированы в 5% случаев спорадических эпилепсий и в 13% семейных форм. Неврологические заболевания, связанные с вариантами в гене DEPDC5, включают семейную фокальную эпилепсию с вариабельными очагами (MIM: 604364), аутосомно-доминантную гипермоторную эпилепсию, связанную со сном, семейную мезиальную и латеральную височную эпилепсию, доброкачественную эпилепсию с центрально-темпоральными спайками и инфантильные спазмы [1][2].
DEPDC5 идентифицирован как один из наиболее распространенных генов эпилепсии, ассоциированных с инфантильными спазмами и внезапной смертью. Хотя при эпилепсии, связанной с DEPDC5, интеллект обычно не страдает, у некоторых людей диагностируют умственную отсталость, расстройство аутистического спектра и другие психиатрические проблемы. Варианты в гене DEPDC5 также были обнаружены у 20% людей с различными аномалиями головного мозга.
Выявление новых вариантов в гене DEPDC5 и исследование их функции необходимо для внедрения прецизионной терапии с использованием ингибиторов mTOR. Однако необходимы дополнительные исследования, чтобы лучше понять функциональное влияние различных вариантов (особенно миссенс-мутаций или вариантов сайта сплайсинга) и их специфическое участие в эпилептогенезе, а также для разработки и использования прецизионных методов лечения у людей.
Прецизионные методы лечения эпилепсии, ассоциированной с DEPDC5, также откроют путь к новым терапевтическим подходам к эпилепсии (включая приобретенные эпилепсии, при которых происходит активация mTORC1, например посттравматическую эпилепсию) и другим неврологическим расстройствам, связанным с дисфункцией пути mTOR [1][2].
В статье представлено два случая семейной фокальной эпилепсии, ассоциированной с вариантом нуклеотидной последовательности в гене DEPDC5.
В психоневрологическом отделении № 1 ГБУЗ «Научно-практический центр специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого ДЗМ» наблюдалась девочка 10 лет с диагнозом «фокальная эпилепсия».
Анамнез жизни
Первый ребенок в неродственном браке, от первой беременности, протекавшей без особенностей. Масса тела при рождении 3850 г, рост 48 см, оценка по шкале Апгар 8/9. Росла и развивалась без особенностей. Наследственность по эпилепсии отягощена: у отца эпилепсия.
Анамнез заболевания
Наблюдается неврологами с возраста 6 лет (с сентября 2018 г.), когда в течение 3 мес с частотой около 2 раз в неделю возникали фокальные приступы: билатеральные тонические с неверсивным поворотом головы и глаз влево, длительностью 1–2 мин. Затем возникла спонтанная ремиссия, в связи с чем противосудорожная терапия не проводилась. С ноября 2021 г. приступы возобновились, приобрели билатеральный тонико-клонический характер. Девочка госпитализирована в психоневрологическое отделение. В терапию введен лаксамид в дозе 150 мг/сут, приступы более не повторялись.
Неврологический статус
На момент осмотра в возрасте 10 лет лицевых и скелетных дисморфий нет. В неврологическом статусе очаговой симптоматики нет, развитие соответствует возрасту.
МРТ
На магнитно-резонансной томографии (МРТ) головного мозга (напряженность магнитного поля 3 Тл) в возрасте 10 лет структурной патологии не выявлено (рис. 1).
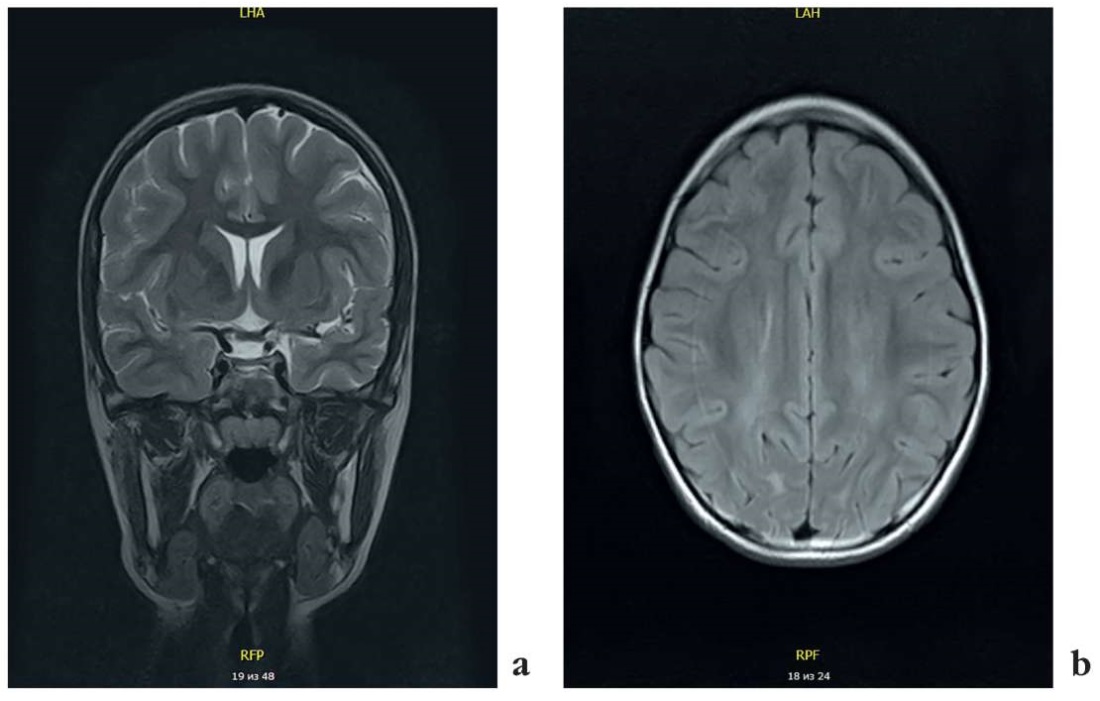
Рисунок 1. Магнитно-резонансная томография головного мозга
пробанда 1 (девочка, 10 лет): без патологии (a, b)
Figure 1. Proband 1 (girl, 10 years old):
brain magnetic resonance imaging, no pathology (a, b)
Видео-ЭЭГ-мониторинг
При проведении видеоэлектроэнцефалографического (видео-ЭЭГ) мониторинга в возрасте 10 лет зарегистрированы периодическое региональное дельта-замедление и региональная эпилептиформная активность в левой лобно-центральной области с заинтересованностью вертексных отделов, низким индексом. Выявлен субклинический ЭЭГ-паттерн фокального приступа, исходящий из F3–C3–Fz–Cz, длительностью 30 с (рис. 2).
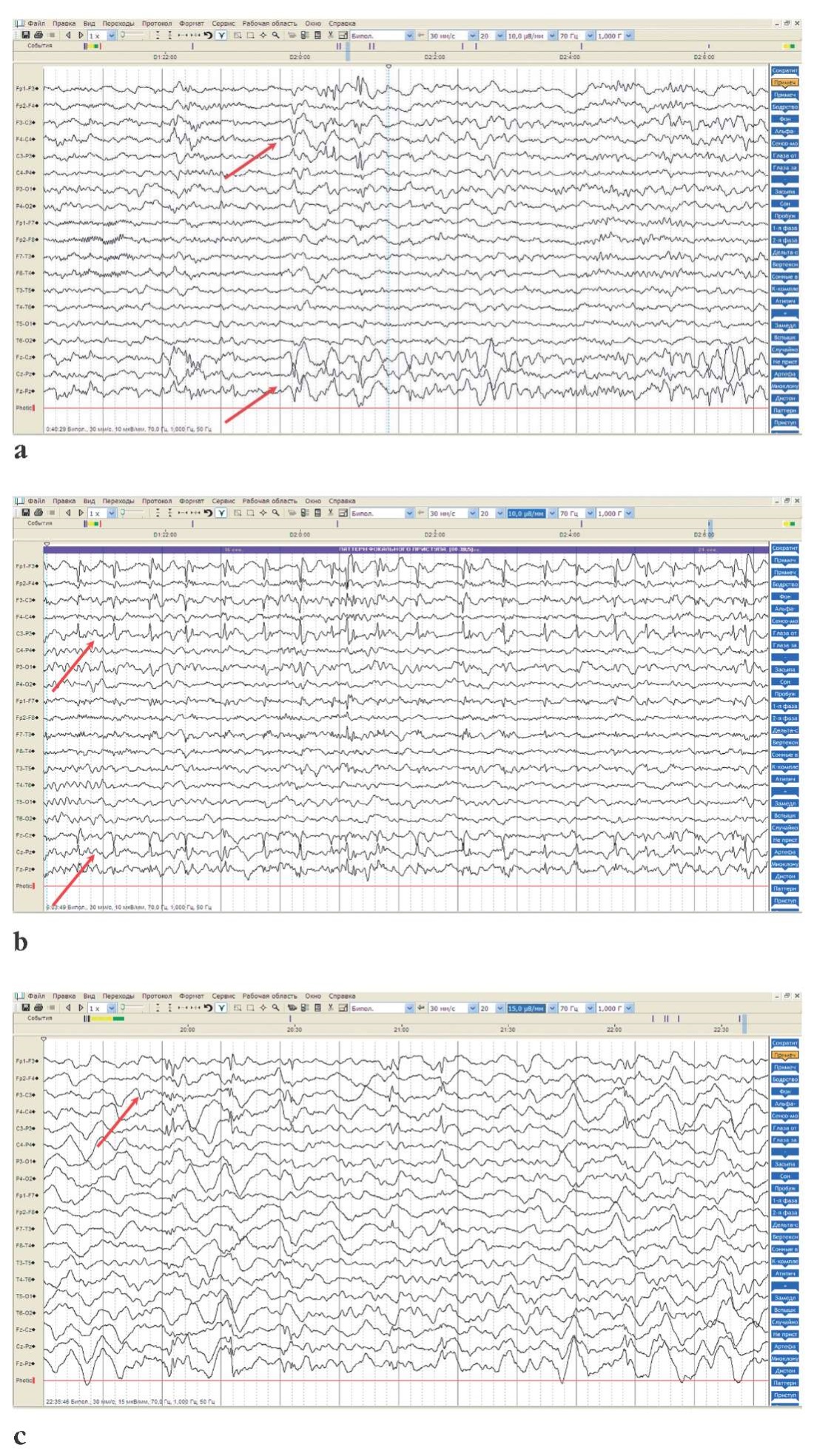
Рисунок 2. Данные видеоэлектроэнцефалографического (ЭЭГ) мониторинга
пробанда 1 (девочка, 10 лет):
а, b – региональное дельта-замедление и региональная эпилептиформная активность
в левой лобно-центральной области (стрелки)
с заинтересованностью вертексных отделов, низким индексом;
с – зарегистрирован субклинический ЭЭГ-паттерн фокального приступа,
исходящий из F3–C3–Fz–Cz, длительностью 30 c (стрелка)
Figure 2. Proband 1 (girl, 10 years old):
video electroencephalographic (EEG) monitoring data:
a, b – regional delta slowing and regional epileptiform activity
in the left fronto-central region (arrows)
involving the vertex regions, with low index;
c – a subclinical focal seizure-related 30 s-long EEG pattern
was recorded from F3–C3–Fz–Cz (arrow)
Генетическое исследование
При информированном согласии родителей проведено полноэкзомное секвенирование. Геномная ДНК выделена методом лизиса клеток с последующей очисткой на стекловолоконных фильтрах (реактивы QIAamp DNA Mini Kit – Qiagen, Нидерланды), затем использована для приготовления геномных библиотек для массового параллельного секвенирования (NEBNext Ultra II – New England BioLabs, США). Из полученных библиотек методом гибридизации были отобраны только те участки ДНК, которые соответствуют экзонам генов и сайтам сплайсинга (SureSelect AllExon V7 – Agilent, США). Далее было проведено определение их нуклеотидной последовательности на секвенаторе HiSeq 1500 (Illumina, США) с использованием реактивов HiSeq Rapid SBS Kit v2. Выявлен гетерозиготный вариант нуклеотидной последовательности в интроне 14 (сайт сплайсинга) гена DEPDC5 (c.946+1G>A, NM_014662.5). Проведена ДНК-диагностика методом секвенирования по Сэнгеру, данный вариант в гене DEPDC5 подтвержден у пробанда и выявлен у отца (рис. 3).
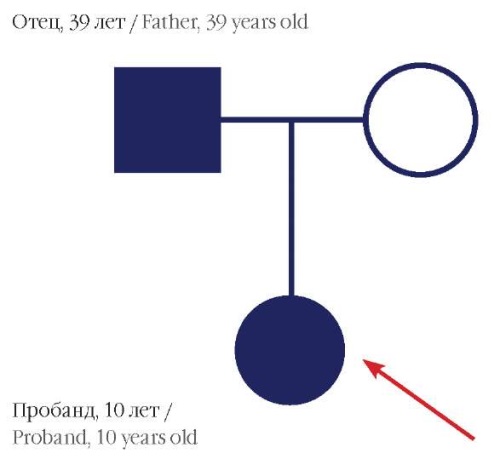
Рисунок 3. Родословная пробанда 1 (указан стрелкой)
Figure 3. Ancestry of proband 1 (indicated by an arrow)
Анамнез отца
Отец пробанда 1983 г.р. наблюдается неврологом с диагнозом «фокальная эпилепсия» с 5 лет. Характер приступов: фокальные с автоматизмами, билатеральные тонико-клонические с фокальным началом. Течение эпилепсии фармакорезистентное.
В течение заболевания получал антиэпилептические препараты (АЭП): фенобарбитал, карбамазепин, вальпроевую кислоту, фенитоин, ламотриджин, топирамат, окскарбазепин, перампанел, прегабалин, зонисамид, лакосамид – без устойчивого положительного эффекта. Наиболее эффективные АЭП: карбамазепин, лакосамид.
С учетом фармакорезистентного течения пациент в 31 год был обследован как кандидат на хирургическое лечение эпилепсии. По данным видео-ЭЭГ-мониторинга регистрировалась эпилептиформная активность в правой и левой височных областях (доминантный очаг справа). По результатам МРТ головного мозга: изменение сигналов в Т1-режиме от обоих гиппокампов, стертость границ по типу фокальной кортикальной дисплазии (ФКД), нарушение строения извилин в базальных отделах левой височной доли по типу полимикрогирии (рис. 4).
В возрасте 31 года пациенту были установлены интракраниальные электроды, проведена электрокортикография, зарегистрировано два тонико-клонических приступа с предположительной зоной начала в правом гиппокампе и последующим вовлечением левого гиппокампа. Проведена селективная амигдалогиппокампэктомия справа.
В настоящее время у отца приступы сохраняются 3–5 раз в месяц. Принимает карбамазепин (1200 мг/сут), лакосамид (300 мг/сут). В неврологическом статусе очаговой симптоматики нет, отмечаются мнестические нарушения.
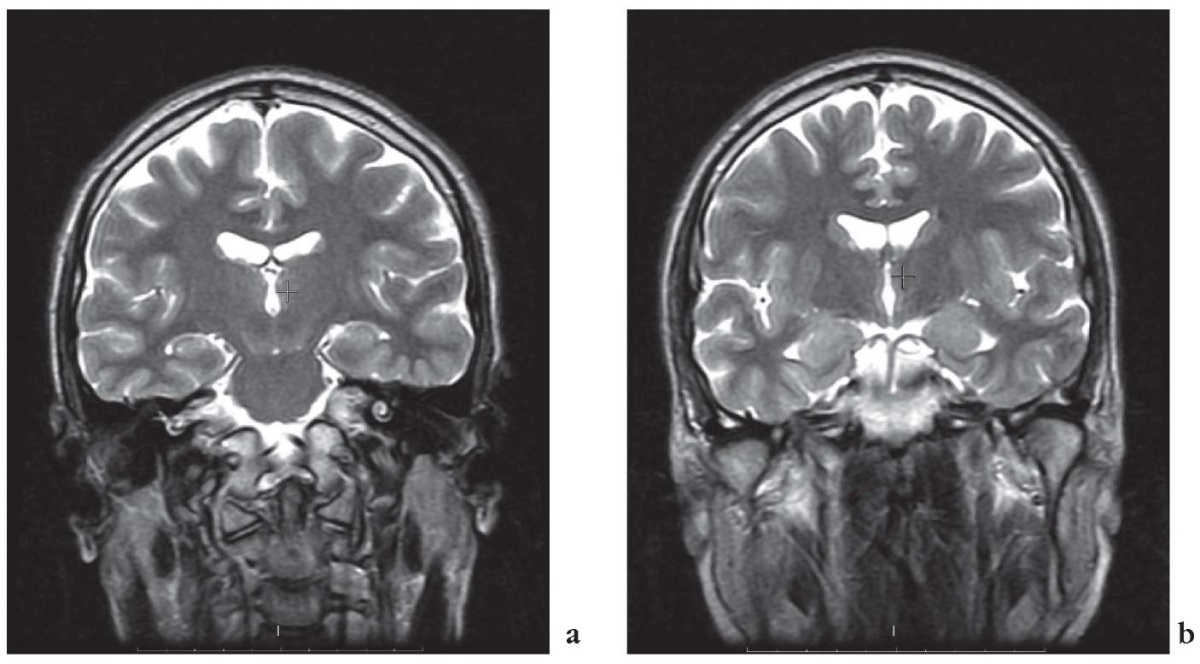
Рисунок 4. Магнитно-резонансные томограммы головного мозга
отца пробанда 1 (до оперативного вмешательства):
изменение сигналов в Т1-режиме от обоих гиппокампов,
стертость границ по типу фокальной кортикальной дисплазии,
нарушение строения извилин в базальных отделах левой височной доли
по типу полимикрогирии (а, b)
Figure 4. Father of Proband 1. Brain magnetic resonance imaging (before surgery):
altered bilateral hippocampus-specific T1-mode signals from both hippocampi,
focal cortical dysplasia-like blurred boundaries,
polymicrogyria-like disrupted gyrus structure
in basal regions of left temporal lobe (a, b)
В психоневрологическом отделении № 1 ГБУЗ «Научно-практический центр специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого ДЗМ» наблюдался мальчик 7 лет с диагнозом «эпилепсия структурная, мультифокальная, фармакорезистентное течение» и когнитивным дефицитом.
Анамнез жизни
Ребенок от второй беременности (первая – гибель плода на 33-й неделе гестации), протекавшей с угрозой прерывания (отслойка плаценты). Роды вторые на 37-й неделе гестации, оперативные. Масса тела при рождении 3380 г, рост 52 см. Психомоторное развитие до 1 года по возрасту.
Анамнез заболевания
Дебют приступов в 1 год 4 мес в виде первых признаков «замирания». Ходит с 1 года.
Начат подбор противосудорожной терапии без значимого эффекта. По данным МРТ головного мозга заподозрена ФКД височной области (структурная незрелость и снижение миелинизации белого вещества субкортикально симметрично в полюсах височных долей, на этом фоне утолщение передних отделов верхней височной извилины слева).
В возрасте 4 лет проведен сеанс радиочастотной диатермокоагуляции по имплантированным электродам в зонах повышенного эпилептогенеза. На фоне лечения отмечено повышение частоты эпилептических приступов. По данным МРТ после оперативного лечения: постманипуляционные кистозно-глиозные изменения в веществе правой теменно-затылочной области. Единичные, мелкие перивентрикулярные кисты и невыраженный перивентрикулярный глиоз, вероятно, в рамках перинатального гипоксического поражения.
По результатам ранее проведенного генетического исследования (селективный метаболический скрининг) – без патологических изменений в показателях концентрации метаболитов.
В связи с отсутствием положительного эффекта от ранее проводимой терапии АЭП ребенок поступил в психоневрологическое отделение для коррекции терапии и кетогенной диеты.
Фенотипические особенности
Внимание неустойчиво, временные представления сформированы. Знает алфавит, цифры, но не читает. Пассивный словарь достаточный. На вопросы отвечает односложно. Фраз не использует. Темп речи замедлен. Интонационную окраску не употребляет. Игровые навыки не сформированы. Контакт формальный. Большую часть времени проводит в одиночестве за телефоном. Периодически бурные протестные реакции. Диффузная мышечная гипотония, походка дискоординированная, шаркающая. Часто запинается, падает. Утром фон настроения снижен. Часто пользуется коляской. Поза расслабленная, голова свисает. Дисметрия.
Видео-ЭЭГ-мониторинг
Выраженное диффузное замедление корковой ритмики. Фоновая ритмика представлена непрерывной активностью всех волновых диапазонов, в фоне доминирует диффузная медленноволновая активность тета-диапазона амплитудой до 300 мкВ. Основной ритм отчетливо не регистрируется. Медленноволновая активность представлена диффузно, невысоким индексом, преимущественно в виде тета-колебаний, по амплитуде не превышающих фоновую ритмику. Высоким индексом регистрируется эпилептиформная активность, представленная комплексами «острая – медленная волна» с амплитудой 400–600 мкВ в виде генерализованных, переднепроекционных и мультирегиональных разрядов, временами имеющих довольно ритмичный характер.
Отмечено множество клинических событий в бодрствовании и во сне в виде вздрагиваний, слабо выраженного напряжения с приведением конечностей, в большинстве случаев не сопровождавшихся изменениями фоновой ритмики. В части случаев наблюдалось появление диффузного умеренно выраженного электродекремента с редукцией фоновой эпилептиформной активности. Вероятнее всего, имеет место сочетание эпилептических приступов (эписпазмы) и пароксизмов неэпилептического характера. В целом картина эпилептической энцефалопатии (рис. 5).
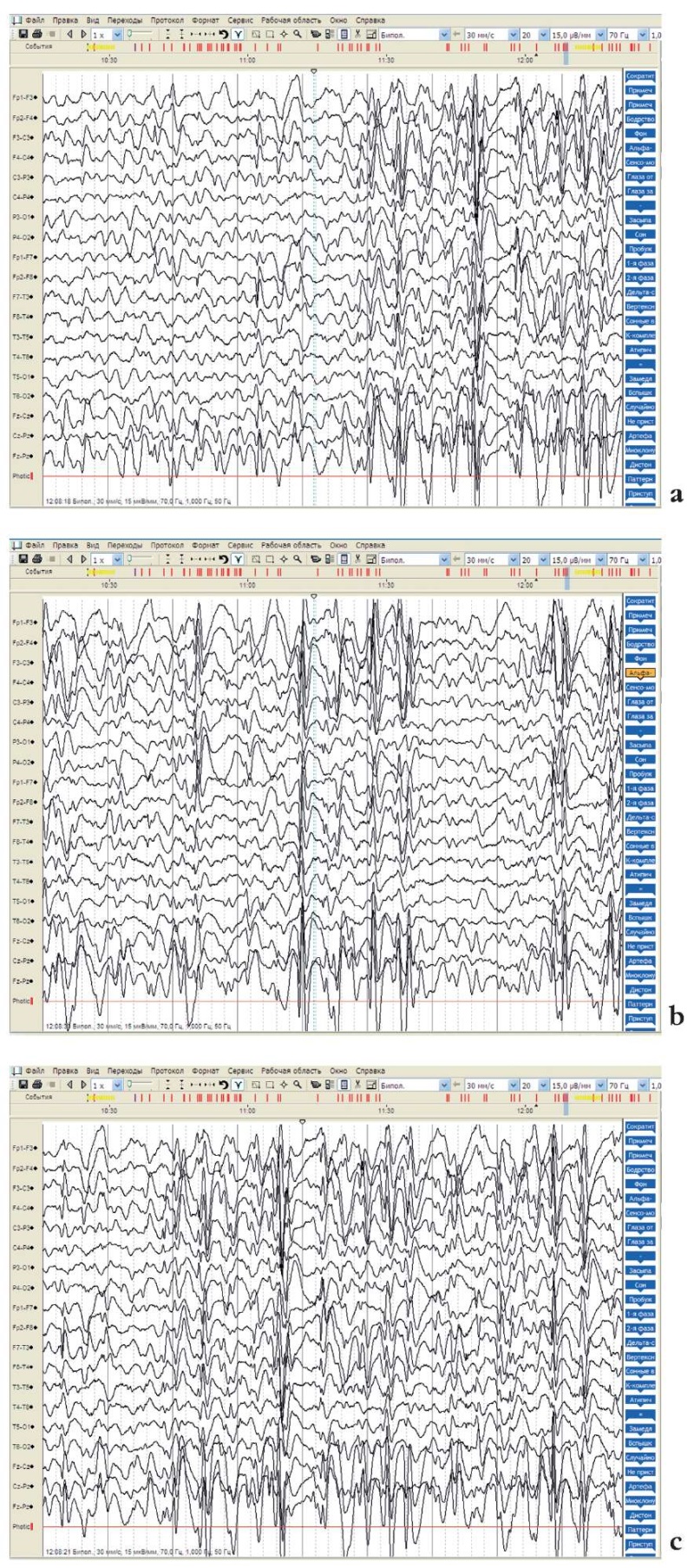
Рисунок 5. Данные видеоэлектроэнцефалографического мониторинга
пробанда 2 (мальчик, 7 лет):
а, b – медленноволновая активность представлена диффузно,
невысоким индексом, преимущественно в виде тета-колебаний,
по амплитуде не превышающих фоновую ритмику;
с – высоким индексом регистрируется эпилептиформная активность,
представленная комплексами «острая – медленная волна» с амплитудой
400–600 мкВ в виде генерализованных,
переднепроекционных и мультирегиональных разрядов,
временами имеющих довольно ритмичный характер
Figure 5. Proband 2 (boy, 7 years old).
Video-electroencephalographic monitoring data:
a, b – diffuse slow-wave activity,
with low index, mainly as theta oscillations,
not exceeding the background rhythm by amplitude;
c – high index, epileptiform activity is recorded,
represented by sharp-slow wave complexes,
with 400–600 μV amplitude, captured as generalized,
anterior projection and multiregional discharges,
periodically showing quite rhythmic pattern
Генетическое исследование
При информированном согласии родителей проведено секвенирование панели генов «Наследственные эпилепсии» (определение их нуклеотидной последовательности на секвенаторе HiSeq 1500 (Illumina, США) с использованием реактивов HiSeq Rapid SBS Kit v2. Выявлен ранее описанный патогенный гетерозиготный вариант нуклеотидной последовательности в экзоне 21 гена DEPDC5 (c.1453C>T; NM_014662.5), приводящий к остановке синтеза полнофункционального белка. Выполнена ДНК-диагностика методом секвенирования по Сэнгеру, данный вариант в гене DEPDC5 подтвержден у пробанда и выявлен у отца. Информация о клиническом состоянии отца неизвестна.
Варианты в гене DEPDC5 ассоциированы с семейной фокальной эпилепсией с вариабельными очагами 1 (англ. familial focal еpilepsy, with variable foci 1) (MIM: 604364). Они передаются по аутосомно-доминантному типу наследования с пенетрантностью от 25% до 100% [3].
К настоящему времени в литературе описано 63 пациента с вариантами в гене DEPDC5 (рис. 6) [2].
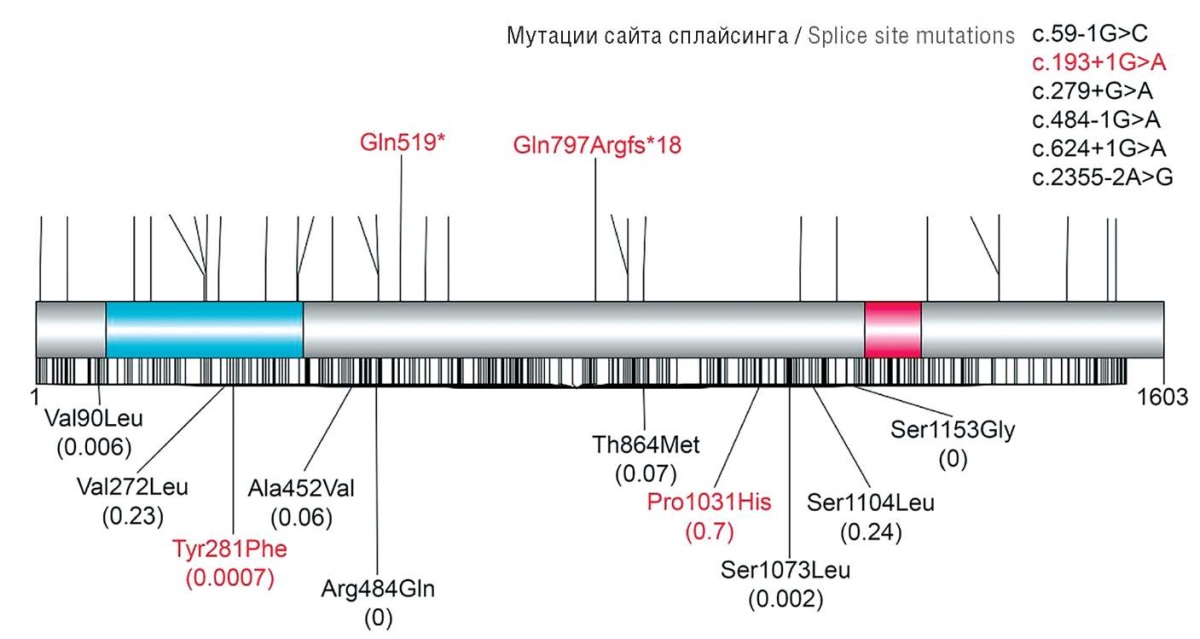
Рисунок 6. Локализация ранее описанных вариантов
нуклеотидной последовательности в гене DEPDC5 [2]
Figure 6. Position of nucleotide sequence variants
previously identified in DEPDC5 gene [2]
Клинический полиморфизм у пациентов с вариантами нуклеотидных последовательностей в гене DEPDC5 разнообразен. Больные, несущие патогенные варианты в сайте сплайсинга и нонсенс-мутации в гене DEPDC5, более подвержены рефрактерной эпилепсии в сочетании с умственной отсталостью, мигренью, расстройством аутистического спектра и психическими нарушениями, в то время как пациенты, несущие патогенные варианты со сдвигом рамки считывания или миссенс-мутации, обычно имеют благоприятный прогноз [2].
Белок, кодируемый геном DEPDC5, является членом комплекса GATOR1, который ингибирует мишени рапамицин-опосредованных процессов у млекопитающих, включая рост и пролиферацию клеток. Имеющиеся данные свидетельствуют, что некоторые мутации, вызывающие эпилепсию, значительно снижают GATOR1-зависимое ингибирование передачи сигналов TORC1. Таким образом, фокальную эпилепсию, связанную с вариантами в гене DEPDC5, классифицируют как «мТОРопатию» [1][4].
В описанном клиническом случае 1 мы наблюдали семью (пробанд и отец) с фокальной эпилепсией, обусловленной вариантом, приводящим к изменению сплайсинга. Клинический фенотип у пробанда включал только фокальные судороги с эпилептиформной активностью в левой лобно-центральной области, без структурных изменений головного мозга, купирующиеся на фоне приема лакосамида, но с сохранением эпилептиформной активности на ЭЭГ. Напротив, у отца пробанда выявлены изменения по типу ФКД и судороги с эпилептиформной активностью в правой и левой височных областях. Возраст начала заболевания у пробанда – 6 лет, у отца – 5 лет, без другой неврологической патологии, с сохранением интеллекта.
В случае 2 определен ранее описанный вариант нуклеотидной последовательности с потерей функции (англ. loss of function), приводящий к остановке синтеза полнофункционального белка [5][6] у пациента с ранним началом приступов, пороком развития коры головного мозга (ФКД височной области), выраженным когнитивным дефицитом и фармакорезистентным течением эпилепсии. В научной литературе также у всех больных с повреждающими вариантами в гене DEPDC5, в первую очередь нонсенс-мутациями, описано более тяжелое течение заболевания и отсутствие положительного эффекта от проводимой противосудорожной терапии [5][6].
У пациентов с эпилепсией, ассоциированной с вариантами в гене DEPDC5, часто наблюдают пороки развития головного мозга и интеллектуальный дефицит. В последние годы группы исследователей описали ФКД у больных эпилепсией с помощью МРТ головного мозга [7][8] и отметили, что большинство случаев ФКД было результатом вторичного соматического варианта в данном гене [9]. Это объясняется тем, что соматические варианты в гене DEPDC5 запускают развитие ФКД через гиперактивацию рапамицина у млекопитающих в дисморфных нейронах [9]. Показана корреляция развития ФКД с рефрактерной эпилепсией с нонсенс-мутациями в гене DEPDC5 [10].
В наших случаях ФКД было выявлена у отца пробанда 1 и у пробанда 2. В обоих наблюдениях ФКД сопровождалась рефрактерной эпилепсией, тогда как у пробанда 1 без ФКД отмечен положительный эффект от применения АЭП.
Представленные клинические случаи подтверждают, что патогенные варианты в гене DEPDC5 ассоциированы с семейной фокальной эпилепсией, клиническое проявление которой может зависеть от типа выявленной мутации. Мы обнаружили у пробанда 1 ранее не описанный гетерозиготный вариант в гене DEPDC5, таким образом расширив спектр мутаций при данной патологии.
Изучение генотип-фенотипических корреляций необходимо для решения вопроса о назначении терапии, в т.ч. и хирургической коррекции, и оценки прогноза у пациентов с эпилепсией, связанной с вариантами в гене DEPDC5. При принятии решения о хирургическом лечении эпилепсии следует проводить генетическое тестирование методом полноэкзомного или полногеномного секвенирования.
1. Samanta D. DEPDC5-related epilepsy: а comprehensive review. Epilepsy Behav. 2022; 130: 108678. https://doi.org/10.1016/j.yebeh.2022.108678.
2. Zhang X., Huang Z., Liu J., et al. Phenotypic and genotypic characterization of DEPDC5-related familial focal epilepsy: case series and literature review. Front Neurol. 2021; 12: 641019. https://doi.org/10.3389/fneur.2021.641019.
3. Tsai M.H., Chan C.K., Chang Y.C., et al. DEPDC5 mutations in familial and sporadic focal epilepsy. Clin Genet. 2017; 92: 397–404. https://doi.org/10.1111/cge.12992.
4. Carvill G.L., Crompton D.E., Regan B.M., et al. Epileptic spasms are a feature of DEPDC5 mTORopathy. Neurol Genet. 2015; 1 (2): e17. https://doi.org/10.1212/NXG.0000000000000016.
5. Dibbens L.M., de Vries B., Donatello S., et al. Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet. 2013; 45 (5): 546–51. https://doi.org/10.1038/ng.2599.
6. Ishida S., Picard F., Rudolf G., et al. Mutations of DEPDC5 cause autosomal dominant focal epilepsies. Nat Genet. 2013; 45 (5): 552–5. https://doi.org/10.1038/ng.2601.
7. Scerri T., Riseley J.R., Gillies G., et al. Familial cortical dysplasia type IIA caused by a germline mutation in DEPDC5. Ann Clin Transl Neurol. 2015; 2 (5): 575–80. https://doi.org/10.1002/acn3.191.
8. Baldassari S., Picard F., Verbeek N.E., et al. The landscape of epilepsyrelated GATOR1 variants. Genet Med. 2019; 21 (2): 398–408. https://doi.org/10.1038/s41436-018-0060-2.
9. De Fusco A., Cerullo M.S., Marte A., et al. Acute knockdown of Depdc5 leads to synaptic defects in mTOR-related epileptogenesis. Neurobiol Dis. 2020; 139: 104822. https://doi.org/10.1016/j.nbd.2020.104822.
10. Gu C., Lu X., Ma J., et al. What is the impact of a novel DEPDC5 variant on an infant with focal epilepsy: a case report. BMC Pediatr. 2022; 22 (1): 459. https://doi.org/10.1186/s12887-022-03515-8.
Кожанова Татьяна Викторовна – к.м.н., ведущий научный сотрудник, врач – лабораторный генетик; доцент кафедры неврологии, нейро-хирургии и медицинской генетики им. академика Л.О. Бадаляна педиатрического факультета
ул. Авиаторов, д. 38, Москва, 119620;
ул. Островитянова, д. 1, Москва, 117997
Жилина Светлана Сергеевна – к.м.н., ведущий научный сотрудник, врач-генетик; доцент кафедры неврологии, нейрохирургии и медицинской генетики им. академика Л.О. Бадаляна педиатрического факультета
ул. Авиаторов, д. 38, Москва, 119620;
ул. Островитянова, д. 1, Москва, 117997
Сушко Лилия Марленовна – врач-невролог
ул. Авиаторов, д. 38, Москва, 119620
Лукьянова Екатерина Геннадьевна – врач-невролог
ул. Авиаторов, д. 38, Москва, 119620
Осипова Карина Вартановна – к.м.н., заведующая психоневрологическим отделением
ул. Авиаторов, д. 38, Москва, 119620
Крапивкин Алексей Игоревич – д.м.н., директор
ул. Авиаторов, д. 38, Москва, 119620
Заваденко Николай Николаевич – д.м.н., профессор, заведующий кафедрой неврологии, нейрохирургии и медицинской генетики им. академика Л.О. Бадаляна педиатрического факультета
ул. Островитянова, д. 1, Москва, 117997
Кожанова Т.В., Жилина С.С., Сушко Л.М., Лукьянова Е.Г., Осипова К.В., Крапивкин А.И., Заваденко Н.Н. DEPDC5-ассоциированная семейная фокальная эпилепсия. Эпилепсия и пароксизмальные состояния. 2023;15(4):339-347. https://doi.org/10.17749/2077-8333/epi.par.con.2023.159
Kozhanova T.V., Zhilina S.S., Sushko L.M., Lukyanova E.G., Osipova K.V., Krapivkin A.I., Zavadenko N.N. DEPDC5-related familial focal epilepsy. Epilepsy and paroxysmal conditions. 2023;15(4):339-347. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2023.159

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































