



Содержание
Перейти к:
https://doi.org/10.17749/2077-8333/epi.par.con.2023.171
Перейти к:
В современной классификации эпилептических синдромов, предложенной Международной Противоэпилептической Лигой в 2022 г., эволюционная и эпилептическая энцефалопатия (англ. developmental and epileptic encephalopathy, DEE), вызванная мутацией в гене KCNQ2, выделена в качестве самостоятельной нозологической формы. Альтернативные названия этого заболевания: DEE 7-го типа или ранняя инфантильная эпилептическая энцефалопатия 7-го типа (OMIM: 613720). В статье представлен краткий обзор литературы по данной теме, а также приведено собственное клиническое наблюдение этой редкой патологии.
Малов А.Г., Калашникова Т.П., Вдовина Н.А. Клинические особенности эволюционной и эпилептической энцефалопатии, вызванной мутацией в гене KCNQ2. Эпилепсия и пароксизмальные состояния. 2023;15(4):354-360. https://doi.org/10.17749/2077-8333/epi.par.con.2023.171
Malov А.G., Kalashnikova Т.P., Vdovina N.А. Clinical features of developmental and epileptic encephalopathy caused by KCNQ2 gene mutation. Epilepsy and paroxysmal conditions. 2023;15(4):354-360. https://doi.org/10.17749/2077-8333/epi.par.con.2023.171
Эволюционная и эпилептическая энцефалопатия (англ. developmental and epileptic encephalopathy, DEE), вызванная мутацией в гене KCNQ2 (KCNQ2-DEE), зарегистрирована в OMIM (англ. Online Mendelian Inheritance in Man) под кодовым номером 613720. Альтернативные названия: DEE 7-го типа или ранняя инфантильная эпилептическая энцефалопатия 7-го типа [1].
Ген KCNQ2 (англ. potassium channel, voltage-gated, KQT-like subfamily, member 2) расположен в локусе 20q13.33 и кодирует потенциал-зависимый калиевый канал, который экспрессируется в головном мозге [2]. Спектр болезней, ассоциированных с различными мутациями в этом гене, включает кроме DEE 7-го типа доброкачественную неонатальную эпилепсию 1-го типа и/или миокимию (OMIM: 121200), что является примером фенотипической аллельной гетерогенности [1]. Оба заболевания наследуются по аутосомно-доминантному типу. KCNQ2-DEE чаще возникает вследствие миссенс-мутаций de novo.
Основными клиническими признаками KCNQ2-DEE являются фармакорезистентные эпилептические судорожные припадки, возникающие в раннем детстве, часто в неонатальном периоде, а также стойкие тяжелые психоневрологические нарушения, которые сохраняются даже при ремиссии приступов [3]. В связи со сходной клиникой припадков и выявлением на электроэнцефалограмме (ЭЭГ) в дебюте паттерна «вспышка – подавление» нередко болезнь рассматривалась как разновидность ранней инфантильной DEE Отахары [4].
В современной классификации эпилептических синдромов, предложенной Международной Противоэпилептической Лигой (англ. International League Against Epilepsy, ILAE) в 2022 г., заболевание выделено в качестве самостоятельной нозологической формы в подгруппе этиологически специфических синдромов, входящих в эпилептические синдромы с началом в неонатальном периоде и младенчестве [5]. Обязательными критериями диагностики KCNQ2-DEE названы тонические, миоклонические и/или другие фокальные припадки в сочетании с паттерном «вспышка – подавление» либо мультифокальными разрядами на ЭЭГ, возникшие до 3-месячного возраста, а также очевидная на момент начала заболевания задержка развития нервной системы или энцефалопатия [6]. В легких случаях гетерозиготные мутации в гене KCNQ2 проявляются фенотипом самокупирующейся (семейной) неонатальной эпилепсии (англ. self-limited (familial) neonatal epilepsy, SeLNE). Для нее характерно возникновение на 1-й неделе жизни фокальных (в основном тонических) припадков с изменением латерализации моторных феноменов как во время приступа (миграция), так и в разных приступах (альтернация). В большинстве случаев приступы прекращаются к 6-недельному или, реже, 6-месячному возрасту.
Для KCNQ2-DEE очень характерна различная степень экспрессивности мутантного гена с фенотипическим разнообразием даже при одинаковых мутациях. Так, в одном наблюдении [7] у матери и сына с гетерозиготной мутацией KCNQ2 и неонатальным дебютом эпилепсии отмечался разный исход. У матери уже в младенчестве возникла ремиссия припадков, а впоследствии наблюдалось нормальное психомоторное развитие. У сына, несмотря на лечение, сформировались эпилептическая энцефалопатия с фармакорезистентными судорогами и психическое недоразвитие с гипотонией и дистонией. В другом случае [3] из 4 членов семьи у матери и младшей дочери фенотип соответствовал типичной доброкачественной неонатальной эпилепсии. Однако у родной сестры матери наблюдались фармакорезистентные припадки до 5 лет и умеренная умственная отсталость во взрослом возрасте. У старшей дочери на 8-й день жизни развились кластеры правосторонних тонических приступов. Полиморфные, резистентные к терапии припадки сохранялись до 7 лет. У нее также была тяжелая умственная отсталость с алалией и спастический тетрапарез, что соответствовало DEE.
S. Weckhuysen et al. [8] сообщили о 8 неродственных пациентах с неонатальной эпилептической энцефалопатией, ассоциированной с KCNQ2. У всех приступы возникли на 1-й неделе жизни, а 2 матери ретроспективно отметили внутриутробные подергивания плода в течение последних 2 мес беременности. В дебюте наблюдались ежедневные фармакорезистентные тонические припадки, но в период от 9 мес до 4 лет частота приступов снижалась, и у большинства они прекратились. Семь пациентов имели глубокие психические нарушения и моторные расстройства, чаще – спастический тетрапарез.
Припадки при KCNQ2-DEE зачастую хорошо контролируются антиконвульсантами, но, несмотря на это, у детей наблюдаются серьезные перманентные психоневрологические нарушения. A.T. Berg et al. [9] провели структурированный онлайн-опрос родителей 39 детей различного возраста с KCNQ2-DEE. Дебют приступов состоялся в первые дни жизни. Во время активной стадии эпилепсии у 35 детей припадки наблюдались ежедневно. Серийное возникновение приступов (3 и более в день) было отмечено у 37 детей, причем у 19 из них – более 10 в день. Наиболее распространенными типами припадков были билатеральные тонико-клонические или, реже, тонические. У большинства пациентов наблюдались тяжелые перманентные расстройства в виде нарушений коммуникации, двигательной функции, использования рук, кормления и др. У 72% были нарушения более чем в двух доменах. Четкой связи между сроком ремиссии по приступам и видом или количеством перманентных нарушений выявлено не было.
Наиболее крупное исследование пациентов с KCNQ2-DEE проведено A. Cossu et al. в 2023 г. [10]. По специально разработанной анкете были собраны данные о 80 пациентах различного возраста (от 4 мес до 43 лет) с мутациями KCNQ2 из 14 стран. Интересно, что у 5 пациентов в возрасте от 2 до 11 лет эпилепсии никогда не было. Основываясь на клинических особенностях, авторы разделили больных старше 2 лет (n=71) на три фенотипа по степени тяжести. Средний возраст между группами был сопоставим. При легкой степени тяжести (42%) дети могли ходить и разговаривать. У них реже отмечалась активная эпилепсия, но чаще диагностировался аутизм. Тяжелый фенотип (27%) характеризовался способностью самостоятельно сидеть и стоять, но не ходить, спастическим мышечным гипертонусом и речью, представленной звуками или слогами. Эпилепсия отмечена почти во всех случаях, а аутизм – редко. При глубокой степени тяжести (31%) наблюдались невозможность держать голову высоко, изменение мышечного тонуса с гипо- и/или гипертонусом и алалия. У всех диагностирована эпилепсия, нередко с ежедневными припадками, а аутизм – ни у кого. У пациентов c глубокой степенью тяжести обнаружены определенные особенности эпилепсии: более высокая частота приступов в первые 10 ч жизни, более частое возникновение эпилептических спазмов и выявление паттерна «вспышка – подавление» уже на первой ЭЭГ. Из 75 обследованных с эпилепсией у 41 пациента в возрасте от 5 мес до 32 лет на момент опроса припадков уже не было. Возраст последнего приступа составил от 2 мес до 10 лет. Среди пациентов с активной эпилепсией у лиц старшего возраста частота приступов была ниже по сравнению с пациентами моложе 6 лет. Изменения при магнитно-резонансной томографии (МРТ) головного мозга были выявлены в 43% (32/75) случаев. Отмечены следующие изменения: гипомиелинизация – в 37% случаев, аномалии белого вещества – в 12%, истончение мозолистого тела – в 28%, корковая атрофия – в 9%, пороки развития коры – в 6%, другие неспецифические аномалии – в 50% [10].
В некоторых работах [11] подчеркивается роль каналов KCNQ2 в процессе развития коры головного мозга. В качестве примера приводится наличие у ребенка с KCNQ2-DEE мальформации коры вследствие аномальной нейрональной миграции в виде гетеротопии серого вещества.
Самым характерным типом эпилептиформной активности на ЭЭГ при KCNQ2-DEE, встречающимся более чем в 60% случаев, является паттерн «вспышка – подавление», который временами может быть асимметричным [6]. В других случаях или позднее могут наблюдаться мультифокальные аномалии, включая спайки и острые волны. Возможно диффузное замедление биоэлектрической активности мозга или ее депрессия в одной гемисфере. Описано появление гипсаритмии [12].
В лечении KCNQ2-DEE обычно применяют блокаторы натриевых каналов (БНК), назначение которых зачастую приводит к ремиссии приступов [12]. Неэффективность фенобарбитала, который часто назначают при неонатальных судорогах, и эффективность БНК, таких как карбамазепин и фенитоин, отмечаются как одна из важных характеристик KCNQ2-DEE [6]. В исследовании A. Cossu et al. [10] из 75 пациентов с эпилепсией 33 человека принимали один антиконвульсант, 30 – два или более, а 12 уже не использовали никаких антиэпилептических препаратов. БНК принимали 40 пациентов, причем 18 – в качестве монотерапии; 10 человек в течение всей жизни следовали кетогенной диете.
Рассматриваются варианты таргетной терапии в зависимости от типа мутации, которые могут быть как с усилением функции белка (англ. gain-of-function, GOF), так и с потерей функции (англ. loss-of-function, LOF), что требует разных терапевтических подходов [13]. По данным F. Miceli et al. [14], отсутствие неонатальных судорог при наличии припадков в детском возрасте является надежным и важным клиническим признаком в пользу GOF-вариантов мутаций гена KCNQ2. При мутациях с усилением функции, таких как KCNQ2 R144, авторами предполагается эффективность амитриптилина, который блокирует каналы, содержащие субъединицы R144Q Kv7.2 и Kv7.3. В случае мутаций, вызывающих эффект потери функции нейрональных калиевых (K+) каналов Kv7.2, клинико-электроэнцефалографическое улучшение отмечается при назначении активатора Kv7 габапентина [15]. Ретигабин, селективно открывающий калиевые каналы KV7.2 и KV7.3, также применяют в качестве прецизионной терапии у пациентов с KCNQ2-DEE [16].
В связи с редкой встречаемостью заболевания и тем, что описаний клинических случаев KCNQ2-DEE в отечественной литературе мы не встретили, приводим собственное наблюдение.
Пациент Х. впервые поступил в неврологическое отделение в возрасте 1,5 мес с жалобами на ежедневные приступы в виде тонического поворота головы, чаще вправо, с напряжением конечностей длительностью до 1 мин.
Вся медицинская помощь оказывалась в полном соответствии со стандартами, порядками и клиническими рекомендациями, а также принципами Хельсинкской декларации Всемирной медицинской ассоциации (Форталеза, Бразилия, 2013 г.). У родителей пациента получено информированное согласие на публикацию. Анонимность больного обеспечена отсутствием персональной информации, в т.ч. изображения, имени, инициалов ребенка и номеров медицинских документов.
Из анамнеза известно, что ребенок от первой беременности, протекавшей на фоне анемии, хронического пиелонефрита и острой респираторной вирусной инфекции в 27 нед у матери, срочных родов. Родоразрешение оперативное в связи с тазовым предлежанием. Оценка по шкале Апгар 8/9 баллов, послед без особенностей. Масса тела при рождении 3490 г, рост 53 см. Наследственность по эпилепсии не отягощена.
Со слов мамы, она заметила приступы со 2–3-го дня жизни. Однако из врачей ей «никто не верил», и она «перестала жаловаться». Впервые ребенок осмотрен неврологом в возрасте 1 мес. В связи с подозрением на «эпилептиформные пароксизмы» было рекомендовано проведение ЭЭГ амбулаторно. На стационарное обследование направлен только через 2 нед.
В отделении зафиксированы серийные адверсивные тонические фокальные моторные припадки в виде поворота головы вправо и/или влево с одновременным напряжением конечностей, иногда с формированием позы асимметричного шейного тонического рефлекса. Изменение латерализации поворота головы могло происходить как во время одного приступа (миграция), так и во время разных припадков (альтернация). В начале приступа могло возникать срыгивание, во время – могли наблюдаться билатеральные миоклонии, в основном в ручках, и нистагм, а в конце – автоматизмы в виде движений языка и/или сглатываний. В конце, а иногда и во время припадка ребенок сильно плакал, что свидетельствовало о сохранности сознания. Длительность приступов обычно составляла несколько минут, но нередко они группировались в кластеры до получаса, которые могли повторяться несколько раз в день. Учитывая серийное течение припадков, с первого дня назначены вальпроаты в виде депакина сиропа с постепенным титрованием дозы с 10 до 40 мг/кг/сут. Уже в начале терапии приступы стали протекать легче, но полной ремиссии удалось достичь только на максимальной дозе.
При объективном неврологическом обследовании выявлена диффузная гипотония с неуверенной опорой. Окружность головы 39 см, большой родничок 2×2 см. Проконсультирован педиатром. Установлен диагноз: хроническое расстройство питания по типу постнатальной гипотрофии 2-й степени (масса тела 4,2 кг). После ультразвукового исследования пилорического отдела желудка проконсультирован хирургом. Установлен диагноз: пилороспазм. В связи с подозрением на наследственную болезнь обмена проконсультирован генетиком, взята кровь для тандемной масс-спектрометрии на спектр аминокислот и ацилкарнитинов, результат которой, полученный позднее, оказался без особенностей. Уровень лактата в крови составил 2,7 ммоль/л (при норме 0,5–2,2 ммоль/л).
На ЭЭГ, записанной в дневном сне, зарегистрирована мультирегиональная эпилептиформная активность (ЭА) в виде комплексов «острая – медленная волна» (ОМВ) с четким акцентом в левой лобной области (рис. 1). На повторной ЭЭГ, записанной в дневном сне через 10 дней на дозе депакина 30 мг/кг/сут, отмечена положительная динамика в виде уменьшения амплитуды и представленности ЭА.
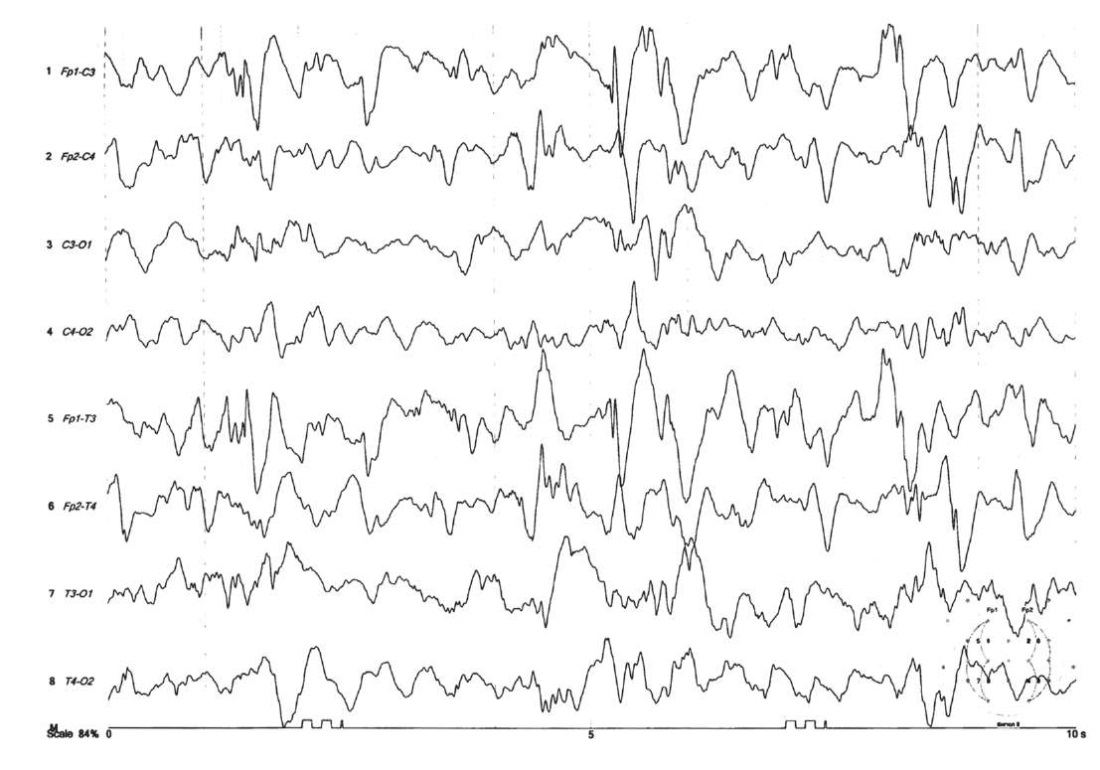
Рисунок 1. Мультирегиональная эпилептиформная активность
с акцентом в левой лобной области
на электроэнцефалограмме пациента Х. (возраст 1,5 мес)
Figure 1. Рatient Х. (aged 1.5 months).
Electroencephalogram-specific multiregional epileptiform activity
highlighting the left frontal region
При МРТ головного мозга данных за наличие изменений очагового и диффузного характера в веществе мозга не выявлено. Пациент был выписан под наблюдение невролога с диагнозом: криптогенная фокальная эпилепсия. Рекомендованы постоянный прием вальпроевой кислоты в дозе 40 мг/кг/сут и лабораторное генетическое обследование для уточнения диагноза.
Повторно поступил на стационарное обследование в возрасте 8 мес с жалобами на задержку психомоторного развития: не поворачивается, не ползает, не сидит. Припадки не повторялись. При объективном обследовании отмечена малая окружность головы (42 см), большой родничок закрыт (постнатальная микроцефалия).
В неврологическом статусе: за предметами не следит, непостоянное альтернирующее расходящееся косоглазие, при тракции за ручки не подтягивается, мышечный тонус повышен по спастическому типу преимущественно в ручках, симптом Бабинского с двух сторон. Офтальмологом впервые отмечена частичная атрофия дисков зрительных нервов с двух сторон.
На ЭЭГ выявлены единичные комплексы ОМВ, но в двух регионах: в левых височных и правых лобно-теменных отведениях. При хромосомном микроматричном анализе патологии не выявлено.
Уже после выписки из отделения были получены результаты секвенирования ДНК по панели «Наследственные болезни обмена веществ» (лаборатория «Геномед», Россия), кровь для которого была взята еще в возрасте 5 мес. Обнаружена «вероятно патогенная мутация, являющаяся возможной причиной заболевания». Выявлена ранее не описанная гетерозиготная мутация в экзоне 17 гена KCNQ2 (chr20:62038349C>CCGTA), приводящая к сдвигу рамки считывания начиная с кодона 756 (p.Gly756fs, NM_172107.2) и нарушающая синтез полноразмерного белка. Также определена гетерозиготная «мутация с неизвестным клиническим значением, имеющая возможное отношение к фенотипу» в гене DHCR7. Гомозиготные мутации в этом гене приводят к развитию аутосомно-рецессивного заболевания – синдрома Смита–Лемли–Опица.
Хотя в заключении было указано, что гетерозиготные мутации в гене KCNQ2 описаны у пациентов с ранней младенческой эпилептической энцефалопатией 7-го типа (OMIM: 613720), а клиническая картина пациента полностью соответствовала ранее описанным проявлениям аутосомно-доминантного заболевания KCNQ2-DEE, генетиком было принято решение о необходимости секвенирования экзома или генома. Результаты полноэкзомного секвенирования ДНК ребенка подтвердили наличие мутаций в обоих генах. Секвенирование этих генов по Сэнгеру у родителей выявило наличие гетерозиготной мутации в гене DHCR7 у матери. Таким образом, мутация в гене KCNQ2 возникла у ребенка de novo.
В возрасте 2 года ребенок стал следить за яркими предметами, удерживать (но не брать) предметы руками, но сидеть и стоять не мог. Хотя на фоне регулярной антиэпилептической терапии (вальпроевая кислота в дозе 30 мг/кг/сут) припадки не повторялись и на ЭЭГ выявлялись только «элементы» ЭА, перманентные нарушения были значительными. Выявлялось психическое недоразвитие с тотальной алалией, сформировался выраженный центральный тетрапарез.
В возрасте 3 года в положении лежа на животе мальчик стал приподниматься на локтях, иногда ползать по-пластунски, переворачиваться, но речи не было. На МРТ головного мозга впервые отмечены признаки неярко выраженной заместительной гидроцефалии как проявления кортикальной атрофии.
При последней госпитализации в возрасте 5 лет положительной динамики в неврологическом статусе не отмечено, навыки сидения и стояния не сформировались. Однако на фоне приема прежней дозы вальпроатов припадки не повторялись и ЭА на ЭЭГ не регистрировалась.
Анализируя картину нашего клинического наблюдения в сравнении с данными литературы, можно отметить следующие особенности. Дебют заболевания зарегистрирован в первые дни жизни, что типично для KCNQ2-DEE [3]. Серии адверсивных припадков с напряжением конечностей и изменением латерализации моторных феноменов, с явлениями миграции и альтернации более характерны для SeLNE [6]. Наблюдалась мультирегиональная эпилептиформная активность на ЭЭГ, хотя более типичным для KCNQ2-DEE считается паттерн «вспышка – подавление» [6]. Отмечен быстрый и стойкий эффект вальпроевой кислоты, блокирующей как кальциевые, так и натриевые потенциал-зависимые каналы, а эффективность БНК – один из важных признаков KCNQ2-DEE, в отличие от других DEE [6][12]. Изменения на МРТ головного мозга в начале заболевания отсутствовали, признаки кортикальной атрофии появились только в возрасте 3 лет, что нередко встречается при KCNQ2-DEE [10]. Выраженные перманентные психоневрологические нарушения, сохранявшиеся даже в возрасте 5 лет, соответствуют глубокой степени тяжести, которая наблюдалась почти у 1/3 пациентов, обследованных A. Cossu et al. [10].
В последние годы установлена корреляция «генотип – фенотип» для KCNQ2-ассоциированных эпилепсий [16]. Гетерозиготные варианты с потерей функции (варианты сдвига рамки считывания, нонсенс-варианты, варианты сплайсинга и некоторые миссенс-варианты) вызывают SeLNE. Миссенс-варианты с доминантно-негативным эффектом соответствуют KCNQ2-DEE, уменьшая калиевые токи в нейронах более чем на 50%. Интересным феноменом является возможность формирования фенотипов KCNQ2-DEE или SeLNE у разных представителей одной семьи [3][7], т.е. при одинаковых мутациях в гене KCNQ2.
Несомненно, что выяснение причин различной степени экспрессивности мутантного гена будет способствовать дальнейшей разработке методов прецизионной терапии KCNQ2-DEE, что активно происходит в последние годы при моногенных эпилептических синдромах [17].
Таким образом, особенностью представленного наблюдения является сочетание признаков KCNQ2-DEE и SeLNE, которая также чаще обусловлена мутациями в гене KCNQ2. Дебют серийных тонических фокальных припадков в сочетании с региональной интериктальной ЭА на ЭЭГ с первых дни жизни, их уменьшение при назначении антиконвульсантов, блокирующих натриевые каналы, возможны при обоих заболеваниях. Изменение латерализации моторных феноменов как во время припадка (миграция), так и в разных приступах (альтернация) более характерно для SeLNE. Однако наличие стойких психоневрологических нарушений, которые сохраняются, несмотря на ремиссию приступов и нормализацию ЭЭГ, наблюдается только при KCNQ2-DEE.
1. OMIM ® . An Online Catalog of Human Genes and Genetic Disorders. URL: https://omim.org (дата обращения 28.09.2023).
2. Biervert C., Schroeder B.C., Kubisch C., et al. A potassium channel mutation in neonatal human epilepsy. Science. 1998; 279 (5349): 403–6. https://doi.org/10.1126/science.279.5349.403.
3. Borgatti R., Zucca C., Cavallini A. et al. A novel mutation in KCNQ2 associated with BFNC, drug resistant epilepsy, and mental retardation. Neurology. 2004; 63 (1): 57–65. https://doi.org/10.1212/01.wnl.0000132979.08394.6d.
4. Saitsu H., Kato M., Koide A., et al. Whole exome sequencing identifies KCNQ2 mutations in Ohtahara syndrome. Ann Neurol. 2012; 72 (2): 298–300. https://doi.org/10.1002/ana.23620.
5. Zuberi S.M., Wirrell E., Yozawitz E., et al. ILAE classification and definition of epilepsy syndromes with onset in neonates and infants: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022; 63 (6): 1349–97. https://doi.org/10.1111/epi.17239.
6. Блинов Д.В. Эпилептические синдромы: определение и классификация Международной Противоэпилептической Лиги 2022 года. Эпилепсия и пароксизмальные состояния. 2022; 14 (2): 101–82. https://doi.org/10.17749/2077-8333/epi.par.con.2022.123.
7. Dedek K., Fusco L., Teloy N., Steinlein O.K. Neonatal convulsions and epileptic encephalopathy in an Italian family with a missense mutation in the fifth transmembrane region of KCNQ2. Epilepsy Res. 2003; 54 (1): 21–7. https://doi.org/10.1016/s0920-1211(03)00037-8.
8. Weckhuysen S., Mandelstam S., Suls A., et al. KCNQ2 encephalopathy: emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol. 2012; 71 (1): 15–25. https://doi.org/10.1002/ana.22644.
9. Berg A.T., Mahida S., Poduri A. KCNQ2-DEE: developmental or epileptic encephalopathy? Ann Clin Transl Neurol. 2021; 8 (3): 666–76. https://doi.org/.1002/acn3.51316.
10. Cossu A., Lo Barco T., Proietti J., et al. Clinical characteristics of 80 subjects with KCNQ2-related encephalopathy: results from a family-driven survey. Epilepsy Behav. 2023; 142: 109153. https://doi.org/10.1016/j.yebeh.2023.109153.
11. Legros L., Adle-Biassette H., Dozières-Puyravel B., et al. Neuropathology findings in KCNQ2 neonatal epileptic encephalopathy. Seizure. 2022; 99: 36–9. https://doi.org/10.1016/j.seizure.2022.05.008.
12. Kato M., Yamagata T., Kubota M., et al. Clinical spectrum of early onset epileptic encephalopathies caused by KCNQ2 mutation. Epilepsia. 2013; 54 (7): 1282–7. https://doi.org/10.1111/epi.12200.
13. Chokvithaya S., Caengprasath N., Buasong A., et al. Nine patients with KCNQ2-related neonatal seizures and functional studies of two missense variants. Sci Rep. 2023; 13 (1): 3328. https://doi.org/10.1038/s41598-023-29924-y.
14. Miceli F., Millevert C., Soldovieri M.V., et al. KCNQ2 R144 variants cause neurodevelopmental disability with language impairment and autistic features without neonatal seizures through a gain-of-function mechanism. EBioMedicine. 2022; 81: 104130. https://doi.org/10.1016/j.ebiom.2022.104130.
15. Soldovieri M.V., Freri E., Ambrosino P., et al. Gabapentin treatment in a patient with KCNQ2 developmental epileptic encephalopathy. Pharmacol Res. 2020; 160: 105200. https://doi.org/10.1016/j.phrs.2020.105200.
16. Borggraefe I., Wagner M. Precision therapy in KCNQ2-related epilepsy. Neuropediatrics. 2023; 54 (5): 295–6. https://doi.org/10.1055/s-0043-1772667.
17. Zimmern V., Minassian B., Korff C. A review of targeted therapies for monogenic epilepsy syndromes. Front Neurol. 2022; 13: 829116. https://doi.org/10.3389/fneur.2022.829116.
Малов Александр Германович – д.м.н., доцент кафедры неврологии и медицинской генетики
ул. Петропавловская, д. 26, Пермь, 614000
Калашникова Татьяна Павловна – д.м.н., профессор кафедры неврологии и медицинской генетики
ул. Петропавловская, д. 26, Пермь, 614000
Вдовина Наталья Анатольевна – врач-невролог отделения неврологии
ул. Героев Хасана, д. 10А, Пермь, 614010
Малов А.Г., Калашникова Т.П., Вдовина Н.А. Клинические особенности эволюционной и эпилептической энцефалопатии, вызванной мутацией в гене KCNQ2. Эпилепсия и пароксизмальные состояния. 2023;15(4):354-360. https://doi.org/10.17749/2077-8333/epi.par.con.2023.171
Malov А.G., Kalashnikova Т.P., Vdovina N.А. Clinical features of developmental and epileptic encephalopathy caused by KCNQ2 gene mutation. Epilepsy and paroxysmal conditions. 2023;15(4):354-360. https://doi.org/10.17749/2077-8333/epi.par.con.2023.171

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































