



Содержание
Перейти к:
https://doi.org/10.17749/2077-8333/epi.par.con.2023.172
Перейти к:
Синдром MELAS (англ. mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes – митохондриальная энцефаломиопатия, лактатацидоз и инсультоподобные эпизоды) относится к группе митохондриальных заболеваний. Большинство случаев синдрома MELAS связаны с мутацией A3243G в гене MTTL1. Частым клиническим проявлением синдрома являются эпилептические приступы (ЭП), которые отличаются фенотипическим полиморфизмом и устойчивостью к противоэпилептической терапии. Диагностика и терапия эпилепсии у пациентов с синдромом MELAS нередко вызывает трудности. В статье представлен клинический случай взрослого пациента с синдромом MELAS с установленной мутацией A3243G и эпилепсией. Наблюдалось прогрессирующее течение заболевания с развитием выраженных когнитивных нарушений. Первые фокальные ЭП возникли на фоне инсультоподобного эпизода. Далее приступы отмечались спонтанно, с высокой частотой, нередко в виде серий. Фокальные ЭП часто имели стертые полиморфные проявления. Выбор медикаментозной терапии учитывал побочные эффекты противоэпилептических препаратов (ПЭП), включая потенциально негативное воздействие на митохондрии. В диагностике ЭП при синдроме MELAS следует учитывать, что приступы нередко возникают во время инсультоподобных эпизодов, могут иметь стертые полиморфные клинические проявления. Наличие когнитивных нарушений у пациентов осложняет выявление ЭП. Препаратами первой линии должны быть ПЭП с низкой митохондриальной токсичностью.
Теплышова А.М., Глазова М.А., Коновалов Р.Н. Эпилепсия и синдром MELAS: обзор литературы и клиническое наблюдение. Эпилепсия и пароксизмальные состояния. 2023;15(4):361-371. https://doi.org/10.17749/2077-8333/epi.par.con.2023.172
Teplysheva А.М., Glazova М.А., Konovalov R.N. Epilepsy and MELAS syndrome: literature review and clinical observation. Epilepsy and paroxysmal conditions. 2023;15(4):361-371. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2023.172
Митохондриальные заболевания (МЗ) – это группа генетических болезней, характеризующаяся нарушением окислительного фосфорилирования, которое возникает в результате мутаций в генах ядерной ДНК и митохондриальной ДНК (мтДНК) [1].
При МЗ в связи с системным дефектом энергетического метаболизма поражаются наиболее энергозависимые ткани и органы-мишени (головной мозг, скелетные мышцы, миокард, орган зрения, печень, почки, эндокринная система) в различных комбинациях, что приводит к полисистемным нарушениям [2]. Характер и тяжесть клинических проявлений МЗ в основном определяются такими факторами, как тяжесть мутации мтДНК, процентное содержание мутантной мтДНК в конкретных органах и тканях, энергетическая потребность и функциональный резерв органов и тканей, содержащих мтДНК [2].
Одной из наиболее известных форм МЗ является синдром MELAS (англ. mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes – митохондриальная энцефаломиопатия, лактатацидоз и инсультоподобные эпизоды). Как отдельная нозологическая единица он был выделен S.G. Pavlakis et al. в 1984 г. [3].
Синдром MELAS может возникать в результате различных мутаций мтДНК [4]. Большинство случаев (приблизительно 80%) связаны с мутацией A3243G в гене MTTL1, кодирующем транспортную РНК [4–7]. Дебют заболевания чаще приходится на детский и молодой возраст [8]. Клиническое течение вариабельное, преимущественно неуклонно прогрессирующее, приводящее к тяжелой инвалидизации [8]. В настоящее время для установления диагноза синдрома MELAS используются определенные диагностические критерии [9][10].
При синдроме MELAS в первую очередь поражаются нервная система и мышцы [8]. Основные клинические проявления включают инсультоподобные эпизоды (ИПЭ), головную боль, когнитивные нарушения, мышечную слабость, непереносимость физических нагрузок [9–11]. Также могут наблюдаться нарушение слуха, зрения, сахарный диабет, поражение сердца, почек и другие расстройства [10]. Для пациентов с синдромом MELAS характерна низкорослость [10].
Одним из наиболее часто встречающихся неврологических расстройств синдрома MELAS являются эпилептические приступы (ЭП), распространенность которых составляет, по данным разных исследований, от 71% до 96% [11][12]. Приступы могут быть как спонтанными, так и спровоцированными, связанными с развитием ИПЭ и другими метаболическими нарушениями [13][14].
Нередко синдром MELAS манифестирует с ЭП. Проспективное когортное исследование S. Yatsuga et al. (2012 г.) [10], проведенное в Японии и включившее 96 пациентов с синдромом MELAS, подтвердило, что ЭП являются одним из наиболее часто встречающихся начальных симптомов наряду с головной болью и ИПЭ. Приступы наблюдались у 36 из 58 больных (62,1%) в ювенильной подгруппе (возраст дебюта синдрома MELAS – до 18 лет) и у 18 из 38 пациентов (47,4%) во взрослой подгруппе [10]. В исследовании J. Li et al. (2021 г.) [15] из 34 больных у 58,82% ЭП были первыми проявлениями МЗ. Еще более высокие показатели получены X. Yang et al. (2023 г.) [16] – ЭП возникали в дебюте синдрома MELAS у 69,1% пациентов.
Эпилептические приступы при синдроме MELAS характеризуются широкой фенотипической гетерогенностью, что неоднократно подчеркивалось в литературе [12][17]. Описаны как фокальные, так и генерализованные ЭП, которые могут возникать изолированно или сочетаться [18]. Нередко авторы указывают, что у пациентов с синдромом MELAS чаще отмечаются фокальные приступы. Так, H.N. Lee et al. (2016 г.) [12], исследуя педиатрическую группу (22 ребенка и подростка) с синдромом MELAS с установленной мутацией мтДНК A3243G, выявили фокальные приступы у 21 пациента (95,5%), генерализованные – у 7 (31,8%). Похожие результаты получены в работе других авторов с участием больных более широкого возрастного диапазона (от 6 мес до 57 лет). ЭП с фокальным началом наблюдались у 58,82% пациентов, с генерализованным – у 32,35%. В 3 случаях (8,82%) ЭП имели неизвестное начало [15]. Однако в ретроспективном исследовании X. Yang et al. (2023 г.) из 97 пациентов (средний возраст начала заболевания 21 год) ЭП с генерализованным началом зарегистрированы более чем в половине случаев (51,5%) [16].
Зона начала фокальных ЭП, по данным литературы, чаще определяется в задних отделах головного мозга (затылочных, теменных, височных), что согласуется со склонностью к возникновению ИПЭ в этих областях [17][19][20]. Приступы, исходящие из затылочной области головного мозга, в основном проявляются зрительными и глазодвигательными симптомами, также может отмечаться иктальная головная боль, тошнота [21]. При распространении эпилептиформной активности на экстраокципитальные области, что достаточно часто бывает при затылочной эпилепсии, наблюдаются симптомы, характерные для вовлечения височной, теменной или лобной доли. При раннем дебюте синдрома MELAS, когда первыми проявлениями являются ЭП, может возникнуть необходимость проведения дифференциального диагноза с идиопатической затылочной эпилепсией. E. Cesaroni et al. (2009 г.) представили описание 19-летнего пациента с 9-летним анамнезом эпилепсии и приступами, исходящими из затылочной доли и характеризующимися отведением глаз и тоническим поворотом головы [22]. Пациент первоначально наблюдался с диагнозом идиопатической детской затылочной эпилепсии типа Гасто. В дальнейшем был установлен синдром MELAS, выявлена мутация мтДНК A3243G. Данный клинический случай показывает, что затылочные приступы могут быть единственным длительным клиническим проявлением синдрома MELAS. В случаях, когда ЭП проявляются головными болями, зрительными нарушениями, они могут быть ошибочно расценены как цефалгия, характерная для синдрома MELAS. Это требует более детального обследования пациентов, в первую очередь – проведения продолженного видеоэлектроэнцефалографического (видео-ЭЭГ) мониторинга для уточнения генеза пароксизмов.
В литературе имеются сообщения о склонности пациентов с синдромом MELAS к возникновению серийных ЭП и развитию эпилептического статуса (ЭС) [17]. В настоящее время описаны клинические наблюдения больных с ЭС с моторными проявлениями (конвульсивный, фокальный) и бессудорожным ЭС [14][19][23–27]. Нередко указывается, что развитие ЭС может быть связано с ИПЭ [14][17][26]. Возраст, в котором ЭС возникает в дебюте синдрома MELAS, значительно варьирует. R. Ribacoba et al. (2006 г.) описали 4 пациентов в возрасте от 27 до 41 года, у которых синдром MELAS был диагностирован во время впервые в жизни возникшего ЭС [26]. D. Corda et al. (2006 г.) [25] представили 38-летнюю пациентку с синдромом MELAS, у которой в дебюте во время первого ИПЭ наблюдался ЭС сложных парциальных приступов. На электроэнцефалограмме (ЭЭГ) были зарегистрированы мультифокальные и периодические латерализованные эпилептиформные разряды. На компьютерной томограмме головного мозга выявлялись изменения, соответствующие ИПЭ, в правой височно-теменно-затылочной области. Также описаны клинические случаи, когда диагноз синдрома MELAS был установлен у пациентов на шестом десятилетии и в дебюте у них возникал ЭС [28][29]. Таким образом, ЭС у больных с синдромом MELAS может иметь полиморфные проявления, вариабельный возраст возникновения, быть первым проявлением МЗ.
В настоящее время не установлено каких-либо специфических электроэнцефалографических изменений, патогномоничных для синдрома MELAS [14][17]. На ЭЭГ может выявляться замедление основной активности или эпилептиформная активность, представленная региональными, мультирегиональными или генерализованными разрядами [14]. Эпилептиформные разряды чаще всего локализуются в затылочной, теменной, задней височной областях [14]. По данным X. Yang et al. (2023 г.) [16], изменения на ЭЭГ у пациентов с MELAS были представлены в виде патологических медленноволновых колебаний в 90,9% случаев, эпилептиформных разрядов – в 68,2%.
Часто эпилепсия у больных с синдромом MELAS имеет неблагоприятное течение и устойчивость к противоэпилептической терапии. Несколько исследований было посвящено выявлению факторов, влияющих на прогноз. В отдельных работах показано, что может прослеживаться взаимосвязь между течением эпилепсии и возрастом дебюта, а также тяжестью проявлений синдрома. H.N. Lee et al. (2016 г.) [12], исследовавшие педиатрическую группу с синдромом MELAS, получили следующие результаты. В подгруппе с более ранним дебютом эпилепсии (до 8 лет) значительно чаще отмечалась устойчивость к противоэпилептической терапии, чем в группе с поздним началом, хотя существенных различий в начальных проявлениях не было. Наблюдалась корреляция между тяжестью течения эпилепсии и выраженностью проявлений синдрома MELAS. X. Yang et al. [16] установили, что медикаментозно-резистентное течение эпилепсии при MELAS связано с более ранним возрастом начала приступов (p=0,013), более высокими показателями по шкале Рэнкина (p<0,001) и более высоким уровнем лактата в сыворотке крови в покое (p=0,009).
Далее представлено клиническое наблюдение пациента с синдромом MELAS и эпилепсией.
Мужчина, 31 год, правша, рост 162 см, масса тела 38 кг.
Ведение пациента осуществлялось сообразно принципам Хельсинкской декларации Всемирной медицинской ассоциации (Форталеза, Бразилия, 2013 г.). От родителей пациента получено информированное согласие для проведения клинического обследования и продолженного видео-ЭЭГ-мониторинга.
Пациент рожден от 23-летней матери, от первой беременности, протекавшей с осложнениями. С 5-го месяца беременности наблюдались протеинурия, повышение артериального давления. На поздних сроках выявилась гипотрофия плода. Роды проходили со стимуляцией на 8-м месяце беременности. Масса тела при рождении 1 кг 650 г, рост 42 см. Раннее развитие протекало нормально. Черепно-мозговых травм не отмечалось. Семейный анамнез по эпилепсии не отягощен. Мать клинически здорова. Генетическое обследование матери не проводилось.
С подросткового возраста пациента беспокоили периодические головные боли давящего диффузного характера, плохая переносимость физических нагрузок, прогрессирующее нарушение зрения. К 20 годам, в период обучения в университете, интенсивность головной боли значительно повысилась. Пациент стал отмечать снижение концентрации внимания, сложности при запоминании, повышенную утомляемость.
В 23 года возникло состояние, сопровождавшееся головной болью выраженной интенсивности давящего и пульсирующего характера, которая локализовалась преимущественно в левой лобной области, а также зрительными проявлениями (говорил, что видит перед глазами «бегущую строку»). На следующий день присоединилось головокружение, появились расплывчатость зрения, снижение слуха. Пациент был госпитализирован в стационар.
При проведении магнитно-резонансной томографии (МРТ) головного мозга в левой затылочно-височной области определялась обширная зона гиперинтенсивного МР-сигнала в режимах Т2, T2 FLAIR. Изменения были расценены как картина острого ишемического инсульта в левой гемисфере головного мозга. Через 1 нед после возникновения первых симптомов состояние пациента ухудшилось, наросла неврологическая симптоматика, возникли сенсорная афазия, правосторонний монопарез в руке, зарегистрированы фокальные приступы с нарушением сознания.
При повторном проведении МРТ головного мозга наблюдалось расширение зоны повышенного МР-сигнала в режиме T2 FLAIR за счет вовлечения височных, угловой, надкраевой, постцентральной извилин, верхней теменной дольки. В режиме DWI – ограничение диффузии (рис. 1). Инфарктоподобный очаг в левом полушарии большого мозга не соответствовал зонам кровоснабжения отдельных артерий (вовлечены бассейны задней и средней мозговых артерий).
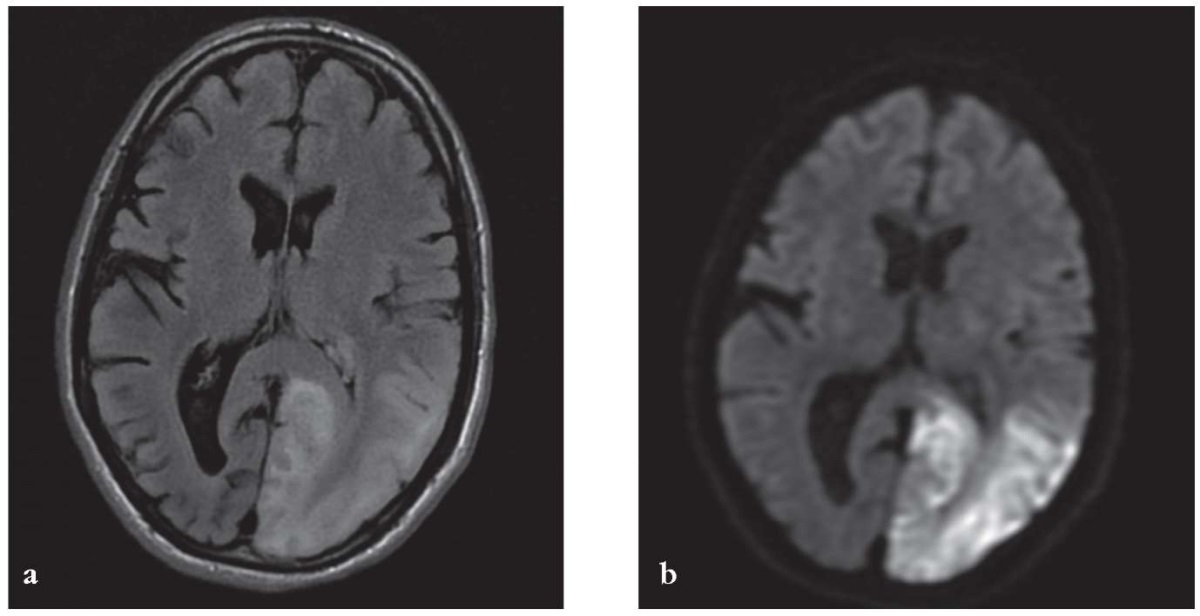
Рисунок 1. Данные магнитно-резонансной томографии головного мозга.
Развитие инсультоподобного эпизода – острый период заболевания.
Инфарктоподобный очаг в левом полушарии большого мозга,
который не соответствует зонам кровоснабжения
(вовлечены бассейны задней и средней мозговых артерий):
a – режим T2 FLAIR, в левом полушарии большого мозга,
в сером и прилежащем белом веществе височной и затылочной долей
выявляется зона повышенной интенсивности сигнала;
b – режим DWI, отмечается ограничение диффузии в очаге
Figure 1. Brain magnetic resonance imaging data.
The development of a stroke-like episode in the acute period of the disease.
An infarct-like focus in the left cerebral hemisphere
not matching to the areas of blood supply
(the basins of the posterior and middle cerebral arteries are involved):
a – T2 FLAIR mode, in the left cerebral hemisphere,
in the gray and adjacent white matter of the temporal and occipital lobes,
an area of increased signal intensity is detected;
b – DWI mode, diffusion limitation in the lesion is noted
На интериктальной ЭЭГ не зарегистрировано эпилептиформной активности.
На фоне проводимой терапии состояние пациента улучшилось. В неврологическом статусе сохранялись правосторонняя гемианопсия, сенсорная афазия, легкая правосторонняя пирамидная недостаточность. Со временем неврологическая симптоматика постепенно регрессировала.
Проведено генетическое исследование. Методом мультиплексной лигазозависимой амплификации (англ. multiplex ligation-dependent probe amplificafion, MLPA) на ДНК, выделенной из клеток крови и мочевого осадка пациента, проведен анализ на частые мутации мтДНК. Выявлена мутация A3243G в клетках крови в гетероплазмическом состоянии (примерно 90% по мутации). Результат подтвержден методом автоматического секвенирования. Также данная мутация обнаружена в гомоплазмическом состоянии на ДНК, выделенной из мочевого осадка пациента.
В анализе крови выявлено повышение лактата до 5,2 ммоль/л.
Установлен диагноз: синдром MELAS.
Через полгода после первого ИПЭ возник повторный с вовлечением правого полушария головного мозга. Во время развития повторного ИПЭ наблюдались следующие клинические проявления: головная боль выраженной интенсивности, дезориентация, возбуждение, левосторонний гемипарез до 4 баллов.
При выполнении МРТ головного мозга в острый период в правой височной области, преимущественно в корковых отделах, определялась зона гиперинтенсивного МР-сигнала в режиме T2 FLAIR без четких контуров. В левой теменно-затылочной области выявлялся участок, соответствующий кистозно-глиозным изменениям с расширением рогов левого бокового желудочка. При динамическом обследовании через 1 нед компьютерная томография головного мозга показала отрицательную динамику в виде увеличения зоны поражения с вовлечением правой височно-теменно-затылочной и частично лобной областей. Восстановление проходило в течение нескольких месяцев.
При проведении МРТ головного мозга через 8 мес после первого ИПЭ выявлена атрофия височных и затылочных долей, выраженное расширение задних рогов боковых желудочков (рис. 2).
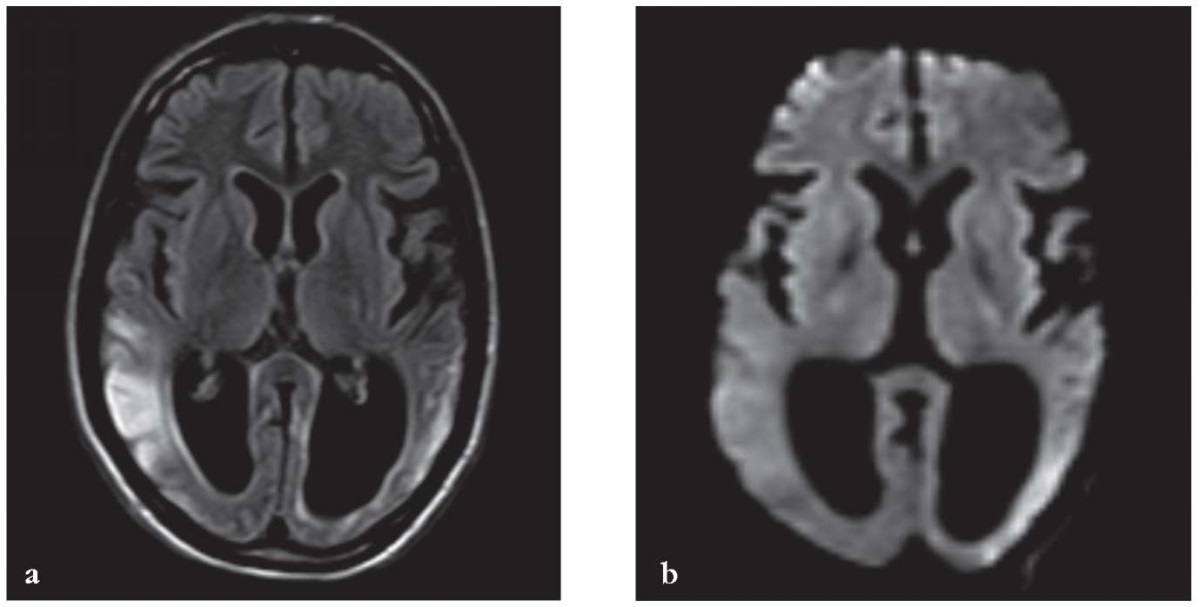
Рисунок 2. Магнитно-резонансные томограммы головного мозга
через 8 мес после первого инсультоподобного эпизода (ИПЭ)
и через 2 мес после второго ИПЭ.
Выявлены атрофия височных и затылочных долей,
выраженное расширение задних рогов боковых желудочков:
a – режим T2 FLAIR, в обоих полушариях большого мозга
в сером и прилежащем белом веществе выявляются
зоны повышенной интенсивности сигнала
на фоне выраженной атрофии вещества мозга;
b – режим DWI, признаков ограничения диффузии не определяется
Figure 2. Brain magnetic resonance imaging
8 months after the first stroke-like episode (IPE)
and 2 months after the second IPE.
Atrophy in the temporal and occipital lobes and prominent expansion
of the posterior horns of the lateral ventricles were revealed:
a – T2 FLAIR mode, in both cerebral hemispheres,
zones of increased signal intensity are detected
in the grey and adjacent white matter
along with prominent atrophy of the brain substance;
b – DWI mode, signs of diffusion limitation are not determined
Пациенту была назначена метаболическая и сосудистая терапия: цитруллин, левокарнитина малат, идебенон, тиоктовая кислота, комплекс поливитаминов и минералов, цитиколин. Отмечена стабилизация состояния, несколько уменьшилась интенсивность головной боли, повторных клинических проявлений ИПЭ не наблюдалось.
Через полгода после второго ИПЭ развился билатеральный тонико-клонический приступ (БТКП) с нарушением сознания, которому предшествовала головная боль выраженной интенсивности. Был назначен леветирацетам в дозе 1000 мг/сут.
С 26 лет пациента стали беспокоить слуховые галлюцинации (музыка, голоса), эти проявления были расценены как психические нарушения, и к схеме лечения добавлен нейролептик кветиопин, который впоследствии заменен на оланзапин. Отмечен частичный положительный эффект.
В связи с развившимся острым аппендицитом, осложненным перитонитом, больному было проведено экстренное хирургическое лечение. При применении миорелаксантов возникло нарушение самостоятельных дыхательных движений вследствие мышечной слабости, в связи с чем в течение 11 сут пациент находился на искусственной вентиляции легких. С этого времени состояние значительно ухудшилось, наросла слабость, стали прогрессировать когнитивные нарушения, расстройства слуха и зрения. Через 1 год был установлен диагноз – сахарный диабет.
С 27 лет возобновились приступы с нарушением сознания, тонико-клоническими судорогами. ЭП преимущественно развивались во сне. Постепенно частота их стала достигать 2–4 раз в месяц, нередко БТКП возникали сериями, 3–4 за ночь. Также появились фокальные приступы, клинически проявляющиеся в виде изменения сознания, прекращения целенаправленной деятельности, могли наблюдаться подергивания в правой руке, автоматизмы слева, при этом пациент не реагировал на обращенную речь, в конце пароксизма мог неадекватно смеяться.
Со временем фокальные ЭП стали проявляться более полиморфно, нередко протекали малозаметно. Продолжительность ЭП составляла около 1 мин, частота точно не известна, примерно 2–6 эпизодов в неделю, нередко в виде серий – около 5 за день. Сам больной на приступы не жаловался в связи с когнитивными нарушениями. Нельзя исключить, что имели место фокальные ЭП без нарушения сознания. Сложности связаны с тем, что нередко было невозможно определить, имеют ли место ЭП или поведенческие нарушения.
Проводилась коррекция противоэпилептической терапии: увеличена доза леветирацетама, затем к терапии добавлен зонисамид. Существенного положительного эффекта не наблюдалось. Продолжали прогрессировать когнитивные нарушения, пациент не мог самостоятельно выполнять привычные действия (одеваться, бриться), перестал различать правую и левую стороны, на вопросы отвечал односложно, нередко невпопад, нарушились способности к счету, чтению. Больной практически перестал проявлять инициативу, временами становился агрессивным.
При проведении повторных МРТ головного мозга отмечено нарастание атрофических процессов в сером и белом веществе височных и затылочных долей.
Впервые пациент был проконсультирован неврологом-эпилептологом летом 2022 г., проводилась коррекция противоэпилептической терапии. С сентября 2022 г. в терапию добавлен ламотриджин, а зонисамид отменен. На фоне приема ламотриджина в дозе 75 мг утром и 125 мг вечером, леветирацетама в дозе 750 мг утром и 1000 мг вечером БТКП были полностью купированы, частота фокальных приступов с нарушением сознания снизилась до 1–2 раз в месяц, они протекали в более легкой форме. Пациент стал активнее. Планируется увеличение дозы ламотриджина.
При проведении видео-ЭЭГ-мониторинга использовали электроэнцефалограф модели Xltek Quantum 256 (Natus, США).
Запись ЭЭГ проводили скальповыми электродами по международной системе «10–20%» и электрокардиографическим (ЭКГ) каналом. При выполнении продолженного видео-ЭЭГ-мониторинга в бодрствовании и во сне зарегистрирована региональная эпилептиформная активность в виде одиночных и сгруппированных комплексов «спайк – медленная волна» в левых теменно-задневисочных отделах с вовлечением затылочной области (рис. 3). Периодически наблюдалось распространение эпилептиформной активности на правую теменно-затылочную область (рис. 4). В бодрствовании и во сне отмечено преходящее билатеральное ритмичное дельта-замедление в вертексно-лобно-центральных отделах полушарий. В ряде случаев указанное замедление приобретало диффузный характер. Во сне также регистрировалось преходящее региональное замедление в правой височной области. В ходе исследования клинических событий, эпилептических приступов и их ЭЭГ-паттернов не выявлено.
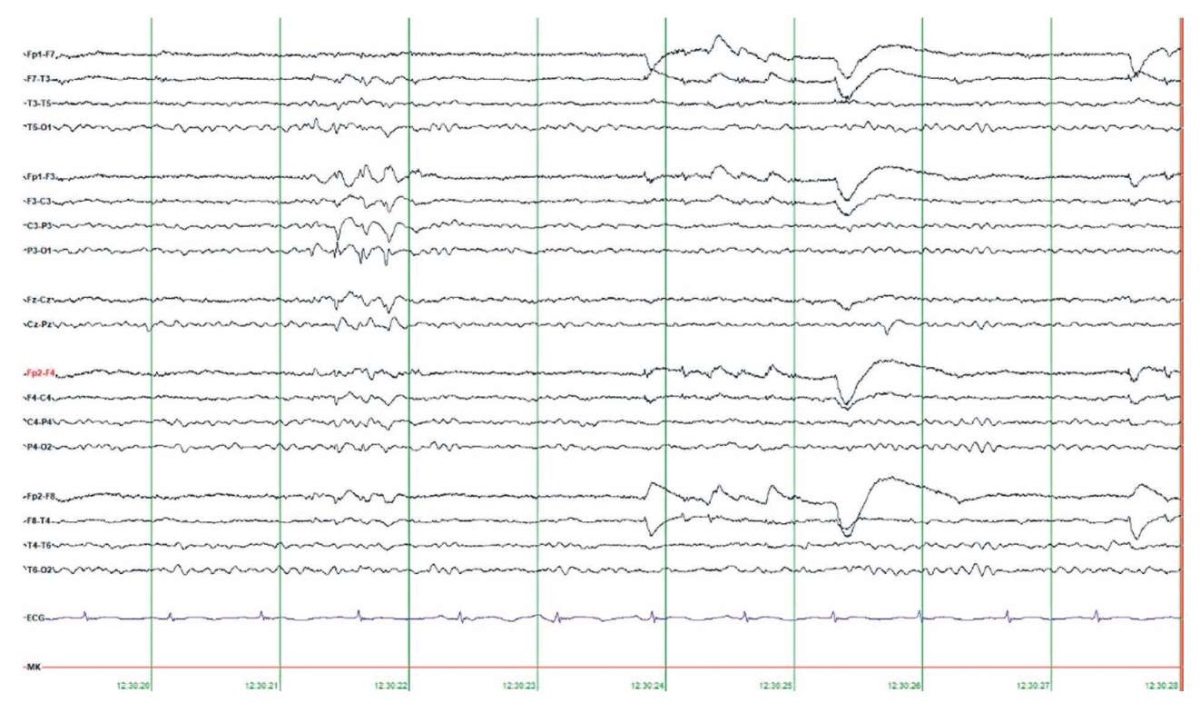
Рисунок 3. Данные электроэнцефалографии, биполярный монтаж.
В бодрствовании зарегистрирована региональная эпилептиформная активность
в виде сгруппированных комплексов «спайк – медленная волна»
в левых теменно-задневисочно-затылочных отделах (P3 – Pz – T5 – O1)
Figure 3. Electroencephalography data, bipolar montage.
During wakefulness, regional epileptiform activity
was recorded as grouped spike-slow wave complexes
in the left parietal-posterior-temporal-occipital regions (P3 – Pz – T5 – O1)
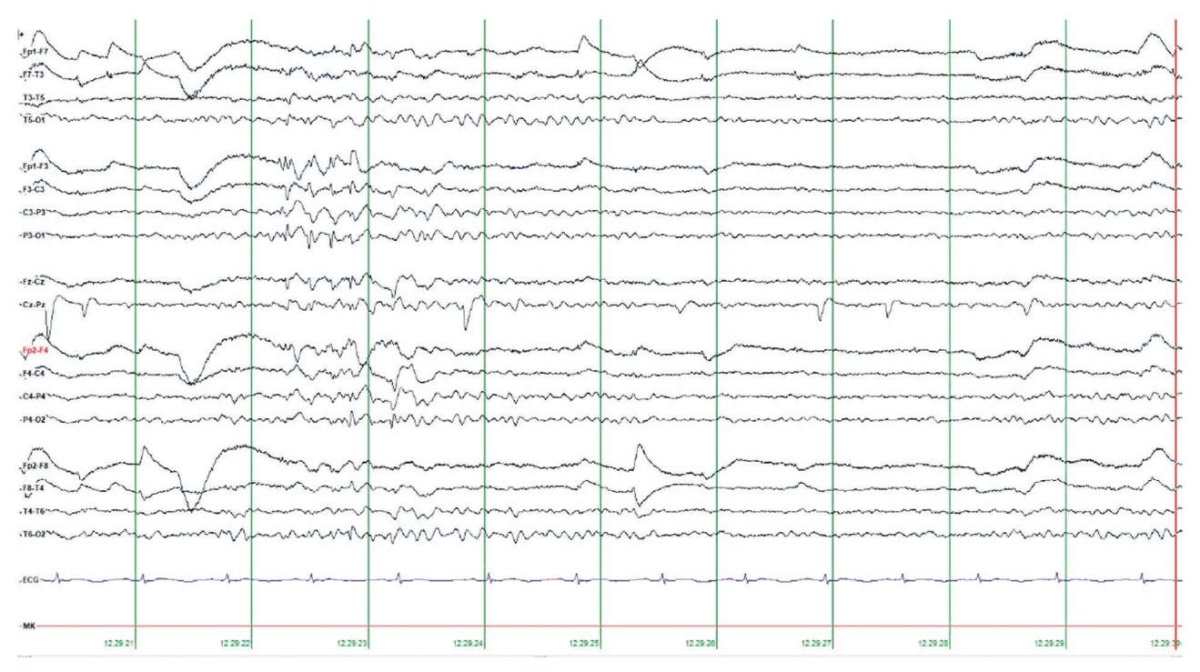
Рисунок 4. Данные электроэнцефалографии, биполярный монтаж.
В бодрствовании зарегистрирована региональная эпилептиформная активность
в виде сгруппированных комплексов «спайк – медленная волна»
в левых теменно-задневисочно-затылочных отделах (P3 – Pz – T5 – O1)
с последующим вовлечением правой теменно-затылочной области (Р4 – О2)
Figure 4. Electroencephalography data, bipolar montage.
During wakefulness, regional epileptiform activity
was recorded as grouped spike-slow wave complexes
in the left parieto-posterior-temporal-occipital regions (P3 – Pz – T5 – O1)
subsequently involving the right parieto-occipital region (P4 – O2)
В представленном клиническом случае дебют синдрома MELAS пришелся на молодой возраст. Одним из первых проявлений был ИПЭ, на фоне которого развились фокальные ЭП. В литературе имеются многочисленные указания на то, что возникновение ИПЭ с развитием ЭП – достаточно типичная картина для синдрома MELAS [10][14][17].
В дальнейшем ЭП у пациента стали возникать спонтанно, имели фокальное начало. Особенностью течения эпилепсии являлась высокая частота приступов, склонность их к серийному течению. Проявления фокальных приступов были полиморфны, нередко малозаметны. Похожие наблюдения представлены в других работах, посвященных пациентам с синдромом MELAS. S.T. Demarest et al. (2014 г.) [17] оценили особенности ЭП и течения эпилепсии у 7 пациентов с мутацией A3243G, ассоциированной с MELAS. Авторы пришли к заключению, что фенотипы эпилепсии разные, однако имеются общие черты: предрасположенность к возникновению продолженных, серийных приступов, унилатеральных моторных ЭП либо приступы могут быть малозаметными, субклиническими.
Особенности течения эпилепсии и проявления приступов ряд авторов объясняет сложным патогенезом, связанным с процессами, происходящими при митохондриальных заболеваниях [14]. Показано, что существует связь между эпилепсией и метаболическими нарушениями в головном мозге [30]. Одной из причин возникновения приступов при МЗ является «истощение» аденозинтрифосфата в результате дефектов окислительного фосфорилирования [14]. Нарушенный митохондриальный метаболизм влечет за собой чрезмерное возбуждение клеток посредством различных механизмов [14]:
Проводимые в последние годы генетические исследования увеличивают число генов, ответственных за развитие эпилепсии при МЗ [14]. Кроме того, у пациентов с синдромом MELAS часто наблюдаются структурные эпилептогенные поражения головного мозга, что имеет дополнительное значение для развития эпилепсии.
В крупном многоцентровом исследовании C. Ticci et al. (2020 г.) [30], проведенном в Италии, при сравнении пациентов с МЗ с ЭП и пациентов без эпилепсии у первых выявлены более высокие показатели аномалий на МРТ, повышенное содержание молочной кислоты в сыворотке крови, а также сопутствующие заболевания. Это подтверждает комплексное влияние различных факторов на развитие ЭП при МЗ.
Еще одной особенностью у описанного нами пациента являлась сложность в выявлении ЭП. Во-первых, это было связано с тем, что фокальные приступы преимущественно не имели выраженных клинических проявлений, нередко протекали малозаметно и их приходилось дифференцировать с поведенческими особенностями. Во-вторых, наличие когнитивных нарушений не позволяло больному самостоятельно оценить и описать пароксизмы. Информация о приступах известна со слов родственников.
Течение эпилепсии у пациентов с MELAS часто носит фармакорезистентный характер, что наблюдалось и в нашем случае. В целом стратегия терапии при синдроме MELAS соответствует общепринятым принципам. Однако, поскольку некоторые противоэпилептические препараты (ПЭП) обладают негативным влиянием на митохондрии, необходимо это учитывать в связи с тем, что их применение может ухудшить состояние больного или даже привести к летальному исходу [31].
ПЭП могут оказывать воздействие на митохондрии путем вмешательства в различные митохондриальные процессы [32]. Они способны влиять на ферментативные каскады, такие как дыхательная цепь или окислительное фосфорилирование, или на пути, не связанные с дыхательной цепью, такие как цикл трикарбоновых кислот или бета-окисление. ПЭП могут воздействовать на каналы, белки-транспортеры, мембранные рецепторы, мембранный потенциал, влиять на механизмы антиоксидантной защиты или нарушать биогенез митохондрий [32].
Наиболее известным ПЭП с выраженной митохондриальной токсичностью является вальпроевая кислота. Еще в 1990-х гг. в нескольких исследованиях была рассмотрена переносимость вальпроатов у пациентов с синдромом MELAS и установлено, что они могут оказывать негативное влияние на митохондрии [33]. Последующие работы подтвердили это [32][34]. Также показано, что вальпроевая кислота способна аггравировать ЭП у пациентов с синдромом MELAS [35]. По данным исследований, митохондриальной токсичностью в разной степени выраженности могут обладать такие ПЭП, как фенобарбитал, карбамазепин, фенитоин, окскарбазепин, этосуксимид, зонисамид, топирамат, габапентин и вигабатрин [32][34]. Однако в настоящее время имеется недостаточное количество исследований, посвященных препаратам последних поколений, в связи с чем оценить их влияние на функции митохондрий не представляется возможным.
В нашем клиническом случае лучшая эффективность наблюдалась при приеме ламотриджина. Кроме того, ламотриджин не оказывал отрицательного влияния на когнитивную и эмоциональную сферы, что позволило стабилизировать психоэмоциональное состояние пациента. Таким образом, адекватное, безопасное, эффективное лечение эпилепсии при синдроме MELAS может улучшить качество жизни больных [12].
Авторы выражают благодарность к.м.н., врачу-неврологу консультативно-диагностического отделения ФГБНУ «Научный центр неврологии» Елене Витальевне Шалимановой за участие в подготовке статьи.
ЭП являются одним из наиболее распространенных клинических проявлений у пациентов с синдромом MELAS. Нередко они возникают в дебюте заболевания и связаны с развитием ИПЭ. Чаще эпилептогенная зона определяется в задних отделах головного мозга (затылочных, теменных, задневисочных), что согласуется со склонностью к возникновению ИПЭ в этих областях.
Эпилепсия у пациентов с синдромом MELAS зачастую резистентна к медикаментозной терапии. Препаратами первой линии в лечении эпилепсии при МЗ должны быть ПЭП, не оказывающие выраженного отрицательного воздействия на функцию митохондрий. Учитывая сложности в диагностике и лечении эпилепсии при МЗ, необходимо продолжать исследования в этой области.
1. Gorman G.S., Chinnery P.F., DiMauro S., et al. Mitochondrial diseases. Nat Rev Dis Prim. 2016; 2 (1): 16080. https://doi.org/10.1038/nrdp.2016.80.
2. Иллариошкин С.Н. Алгоритм диагностики митохондриальных энцефаломиопатий. Нервные болезни. 2007; 3: 23–7.
3. Pavlakis S.G., Phillips P.C., DiMauro S., et al. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome. Ann Neurol. 1984; 16 (4): 481–8. https://doi.org/10.1002/ana.410160409.
4. Chakrabarty S., Govindaraj P., Sankaran B.P., et al. Contribution of nuclear and mitochondrial gene mutations in mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome. J Neurol. 2021; 268 (6): 2192–207. https://doi.org/10.1007/s00415-020-10390-9.
5. Ikeda T., Osaka H., Shimbo H., et al. Mitochondrial DNA 3243A>T mutation in a patient with MELAS syndrome. Hum Genome Var. 2018; 5 (1): 25. https://doi.org/10.1038/s41439-018-0026-6.
6. Goto Y., Horai S., Matsuoka T., et al. Mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS). Neurology. 1992; 42 (3): 545–50. https://doi.org/10.1212/WNL.42.3.545.
7. Finsterer J. Genetic, pathogenetic, and phenotypic implications of the mitochondrial A3243G tRNALeu(UUR) mutation. Acta Neurol Scand. 2007; 116 (1): 1–14. https://doi.org/10.1111/j.1600-0404.2007.00836.x.
8. Pia S., Lui F. Melas syndrome. Treasure Island (FL): StatPearls Publishing; 2023.
9. Hirano M., Ricci E., Koenigsberger M.R., et al. MELAS: an original case and clinical criteria for diagnosis. Neuromuscul Disord. 1992; 2 (2): 125–35. https://doi.org/10.1016/0960-8966(92)90045-8.
10. Yatsuga S., Povalko N., Nishioka J., et al. MELAS: a nationwide prospective cohort study of 96 patients in Japan. Biochim Biophys Acta. 2012; 1820 (5): 619–24. https://doi.org/10.1016/j.bbagen.2011.03.015.
11. El-Hattab A.W., Adesina A.M., Jones J., et al. MELAS syndrome: clinical manifestations, pathogenesis, and treatment options. Mol Genet Metab. 2015; 116 (1–2): 4–12. https://doi.org/10.1016/j.ymgme.2015.06.004.
12. Lee H.N., Eom S., Kim S.H., et al. Epilepsy characteristics and clinical outcome in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS). Pediatr Neurol. 2016; 64: 59–65. https://doi.org/10.1016/j.pediatrneurol.2016.08.016.
13. Lim A., Thomas R.H. The mitochondrial epilepsies. Eur J Paediatr Neurol. 2020; 24: 47–52. https://doi.org/10.1016/j.ejpn.2019.12.021.
14. Lopriore P., Gomes F., Montano V., et al. Mitochondrial epilepsy, a challenge for neurologists. Int J Mol Sci. 2022; 23 (21): 13216. https://doi.org/10.3390/ijms232113216.
15. Li J., Zhang W., Cui Z., et al. Epilepsy associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. Front Neurol. 2021; 12: 675816. https://doi.org/10.3389/fneur.2021.675816.
16. Yang X., Sun A., Ji K., et al. Clinical features of epileptic seizures in patients with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes. Seizure. 2023; 106: 110–6. https://doi.org/10.1016/j.seizure.2023.02.014.
17. Demarest S.T., Whitehead M.T., Turnacioglu S., et al. Phenotypic analysis of epilepsy in the mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes-associated mitochondrial DNA A3243G mutation. J Child Neurol. 2014; 29 (9): 1249–56. https://doi.org/10.1177/0883073814538511.
18. Finsterer J., Zarrouk-Mahjoub S. Focal and generalized seizures may occur in mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS) patients. J Child Neurol. 2015; 30 (11): 1553–4. https://doi.org/10.1177/0883073814567539.
19. Karkare S., Merchant S., Solomon G., et al. MELAS with A3243G mutation presenting with occipital status epilepticus. J Child Neurol. 2009; 24 (12): 1564–7. https://doi.org/10.1177/0883073809334386.
20. Murakami H., Ono K. MELAS: mitochondrial encephalomyopathy, lactic acidosis and stroke-like episodes. Brain Nerve. 2017; 69 (2): 111–7 (in Japanese). https://doi.org/10.11477/mf.1416200650.
21. Мухин К.Ю., Миронов М.Б., Никифорова Н.В. и др. Эпилепсия при синдроме MELAS. Русский журнал детской неврологии. 2009; 4 (3): 8–16.
22. Cesaroni E., Scarpelli M., Zamponi N., et al. Mitochondrial encephalomyopathy lactic acidosis and strokelike episodes mimicking occipital idiopathic epilepsy. Pediatr Neurol. 2009; 41 (2): 131–4. https://doi.org/10.1016/j.pediatrneurol.2009.02.018.
23. Kaufman K.R., Zuber N., Rueda-Lara M.A., et al. MELAS with recurrent complex partial seizures, nonconvulsive status epilepticus, psychosis, and behavioral disturbances: case analysis with literature review. Epilepsy Behav. 2010; 18 (4): 494–7. https://doi.org/10.1016/j.yebeh.2010.05.020.
24. Toribe Y., Tominaga K., Ogawa K., Suzuki Y. Usefulness of L-arginine infusion for status epilepticus in mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes. No To Hattatsu. 2007; 39 (1): 38–43 (in Japanese).
25. Corda D., Rosati G., Deiana G.A., et al. “Erratic” complex partial status epilepticus as a presenting feature of MELAS. Epilepsy Behav. 2006; 8 (3): 655–8. https://doi.org/10.1016/j.yebeh.2005.12.011.
26. Ribacoba R., Salas-Puig J., González C., et al. Characteristics of status epilepticus in MELAS. Analysis of four cases. Neurologia. 2006; 21 (1): 1–11 (in Spanish).
27. Alenezi A.F., Almelahi M.A., Fekih-Romdhana F., et al. Delay in diagnosing a patient with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS) syndrome who presented with status epilepticus and lactic acidosis: a case report. J Med Case Rep. 2022; 16 (1): 361. https://doi.org/10.1186/s13256-022-03613-2.
28. Vrettou C.S., Zervakis D., Priovolos A., et al. MELAS syndrome diagnosed in ICU in a 56-year-old patient presenting with status epilepticus. Intensive Care Med. 2013; 39 (6): 1148–9. https://doi.org/10.1007/s00134-013-2884-1.
29. Leff A.P., McNabb A.W., Hanna M.G., et al. Brief communication complex partial status epilepticus in late-onset MELAS. Epilepsia. 1998; 39 (4): 438–41. https://doi.org/10.1111/j.1528-1157.1998.tb01397.x.
30. Ticci C., Sicca F., Ardissone A., et al. Mitochondrial epilepsy: a cross-sectional nationwide Italian survey. Neurogenetics. 2020; 21 (2): 87–96. https://doi.org/10.1007/s10048-019-00601-5.
31. Finsterer J., Zarrouk Mahjoub S. Epilepsy in mitochondrial disorders. Seizure. 2012; 21 (5): 316–21. https://doi.org/10.1016/j.seizure.2012.03.003.
32. Finsterer J., Scorza F.A. Effects of antiepileptic drugs on mitochondrial functions, morphology, kinetics, biogenesis, and survival. Epilepsy Res. 2017; 136: 5–11. https://doi.org/10.1016/j.eplepsyres.2017.07.003.
33. Lam C.W., Lau C.H., Williams J.C., et al. Mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes (MELAS) triggered by valproate therapy. Eur J Pediatr. 1997; 156 (7): 562–4. https://doi.org/10.1007/s004310050663.
34. Finsterer J., Zarrouk Mahjoub S. Mitochondrial toxicity of antiepileptic drugs and their tolerability in mitochondrial disorders. Expert Opin Drug Metab Toxicol. 2012; 8 (1): 71–9. https://doi.org/10.1517/17425255.2012.644535.
35. Lin C.M., Thajeb P. Valproic acid aggravates epilepsy due to MELAS in a patient with an A3243G mutation of mitochondrial DNA. Metab Brain Dis. 2007; 22 (1): 105–9. https://doi.org/10.1007/s11011-006-9039-9.
Теплышова Анна Михайловна – к.м.н., старший научный сотрудник лаборатории клинической нейрофизиологии
Волоколамское ш., д. 80, Москва, 125367
Глазова Мария Александровна – аспирант 2-го неврологического отделения с лабораторией кардионеврологии
Волоколамское ш., д. 80, Москва, 125367
Коновалов Родион Николаевич – к.м.н., старший научный сотрудник отдела лучевой диагностики
Волоколамское ш., д. 80, Москва, 125367
Теплышова А.М., Глазова М.А., Коновалов Р.Н. Эпилепсия и синдром MELAS: обзор литературы и клиническое наблюдение. Эпилепсия и пароксизмальные состояния. 2023;15(4):361-371. https://doi.org/10.17749/2077-8333/epi.par.con.2023.172
Teplysheva А.М., Glazova М.А., Konovalov R.N. Epilepsy and MELAS syndrome: literature review and clinical observation. Epilepsy and paroxysmal conditions. 2023;15(4):361-371. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2023.172

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































