


Содержание
Перейти к:
 Т. В. Кожанова,
С. С. Жилина,
Т. И. Мещерякова,
Л. М. Сушко,
К. В. Осипова,
А. М. Мазур,
С. С. Фоменко,
А. И. Крапивкин,
Н. Н. Заваденко
Т. В. Кожанова,
С. С. Жилина,
Т. И. Мещерякова,
Л. М. Сушко,
К. В. Осипова,
А. М. Мазур,
С. С. Фоменко,
А. И. Крапивкин,
Н. Н. Заваденко https://doi.org/10.17749/2077-8333/epi.par.con.2024.175
Перейти к:
Варианты нуклеотидной последовательности в гене SEMA6B в большинстве случаев обусловливают развитие фенотипа прогрессирующей миоклонус-эпилепсии и, в меньшей степени, энцефалопатии развития с эпилепсией или без нее. Повреждающие (англ. loss-of-function) варианты нуклеотидной последовательности, локализованные в основном в 17-м экзоне гена, способствуют синтезу аберрантных белков с «токсическими» функциями. В статье описан клинический случай статусного течения эпилепсии у пациента с вариантом в гене SEMA6B (c.2506delС; p.His836ThrfsTer136; NM_032108.4). Представленное наблюдение расширяет наши знания о вариантах в гене SEMA6B, вызывающих прогрессирующую миоклонус-эпилепсию.
Кожанова Т.В., Жилина С.С., Мещерякова Т.И., Сушко Л.М., Осипова К.В., Мазур А.М., Фоменко С.С., Крапивкин А.И., Заваденко Н.Н. SEMA6B-ассоциированная прогрессирующая миоклонус-эпилепсия у пациента с синдромом Клайнфельтера. Эпилепсия и пароксизмальные состояния. 2024;16(1):45-53. https://doi.org/10.17749/2077-8333/epi.par.con.2024.175
Kozhanova T.V., Zhilina S.S., Meshcheryakova T.I., Sushko L.M., Osipova K.V., Mazur A.M., Fomenko S.S., Krapivkin A.I., Zavadenko N.N. SEMA6B-related progressive myoclonus epilepsy in a patient with Klinefelter syndrome. Epilepsy and paroxysmal conditions. 2024;16(1):45-53. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2024.175
Прогрессирующая миоклонус-эпилепсия (ПМЭ; англ. еpilepsy, progressive myoclonic, 11; MIM: 618876) представляет собой группу редких клинически и генетически гетерогенных заболеваний с дебютом в детском или подростковом возрасте и проявляется корковым миоклонусом, генерализованными тонико-клоническими судорогами и прогрессирующими неврологическими нарушениями различной степени тяжести.
В дополнение к описанным в научной литературе случаям ПМЭ, обусловленным различными генетическими нарушениями, последние достижения в области массового параллельного секвенирования позволили идентифицировать дополнительные патогенные варианты в генах, ранее не связанные с фенотипом ПМЭ [1–3]. Среди них повреждающие (англ. loss-of-function) варианты в гене SEMA6B (NM_032108.4), которые впервые были описаны у пяти неродственных пациентов с ПМЭ. Ген SEMA6B локализован в локусе p13.3 на хромосоме 19 и кодирует трансмембранный белок семейства семафоринов, играющий роль в раннем развитии центральной нервной системы [4]. В настоящее время в базах данных и научной литературе описано 29 пациентов с вариабельными фенотипами – от энцефалопатии развития без эпилепсии до энцефалопатии развития и эпилептической энцефалопатии или ПМЭ [1][5–8].
В статье представлен клинический случай тяжелого течения фокальной эпилепсии с синдромом инфантильных спазмов и билатеральными тонико-клоническими судорогами (эпилептическая энцефалопатия) с летальным исходом, ассоциированной с вариантом нуклеотидной последовательности в гене SEMA6B у пациента с полисомией по Х-хромосоме (47,ХХY) – синдромом Клайнфельтера.
В психоневрологическом отделении № 1 ГБУЗ «Научно-практический центр специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого Департамента здравоохранения г. Москвы» (ГБУЗ «НПЦ спец. мед. помощи детям ДЗМ») наблюдался мальчик 2 лет (2021 г.р.) с диагнозом: «Эпилепсия фокальная с синдромом инфантильных спазмов неизвестной этиологии. Атрофия зрительного нерва. Фоновая ретинопатия. Синдром Клайнфельтера (кариотип 47,ХХY). Выраженная задержка психопредречевого и моторного развития.
Ведение пациента осуществлялось сообразно принципам Хельсинкской декларации Всемирной медицинской ассоциации (Форталеза, Бразилия, 2013 г.). От родителей мальчика получено информированное согласие на проведение генетического исследования (полноэкзомного секвенирования).
Ребенок от третьей беременности, третьих родов (в том же браке у родителей две здоровые девочки). Беременность протекала на фоне гестоза в последние месяцы, с 30-й недели – задержка развития плода. На 34-й неделе беременности при кардиотокографии выявлено снижение кровотока. Экстренные оперативные роды на 35-й неделе гестации.
Масса тела при рождении 2040 г, рост 45 см. После рождения в связи с нарушением дыхания ребенок переведен в отделение реанимации и интенсивной терапии (ОРИТ). Мальчик плохо прибавлял в весе, рос и развивался с выраженной задержкой. Взгляд не фиксировал, не плакал и не улыбался. В ГБУЗ «Морозовская детская городская клиническая больница ДЗМ» проведено кариотипирование, установлен синдром Клайнфельтера (47,XXY).
В возрасте 8 мес (05.07.2022 г.) впервые отмечен асимметричный тонико-клонический приступ с поворотом головы и глаз вправо длительностью до 2 мин; 15.07.2022 г. приступ повторился. С помощью электроэнцефалографии (ЭЭГ) выявлена эпилептиформная активность, назначена вальпроевая кислота.
При проведении видео-ЭЭГ-мониторинга (09.08.2022 г.) в бодрствовании и во сне отмечены продолженное региональное дельта-замедление, тета-замедления как независимо в правой и левой затылочных областях, так и билатерально. Низким индексом регистрировалась мультирегиональная эпилептиформная активность в структуре регионального замедления в правой и левой затылочных областях, правой центральной области.
Во время нахождения в стационаре ГБУЗ «Детская городская клиническая больница № 9 им. Г.Н. Сперанского ДЗМ» (ДГКБ № 9) получал курс дексаметазона по 0,2 мл 2 раза в сутки внутримышечно № 4 (20–26.08.2022 г.).
В возрасте 10 мес (13.09.2022 г.) на фоне повышения температуры наблюдалась серия приступов по типу эпилептических спазмов (по описанию матери) в течение 20 мин, купированы введением диазепама; 24.09.2022 г. вновь возник приступ в виде тонического напряжения, заведения глаз вверх с последующими эпилептическими спазмами, купирован самостоятельно.
Пациент постоянно принимал вальпроевую кислоту в дозе 660 мг/сут. Приступы продолжались. В возрасте 10 мес для коррекции противосудорожной терапии ребенок госпитализирован в психоневрологическое отделение № 1 ГБУЗ «НПЦ спец. мед. помощи детям ДЗМ».
Неврологический статус
Судорог на момент осмотра нет. Большой родничок (1×1 см). Язык во рту по средней линии, стереотипное сосание языка и нижней губы. Срыгивает. Сухожильные рефлексы снижены, D=S. Выраженная задержка психомоторного развития: не держит голову, не переворачивается, при тракции за руки не группируется, опоры на ноги нет, других моторных навыков нет, взгляд фиксирует кратковременно, не прослеживает, улыбается редко, гуление скудное.
В связи с ранним началом и прогрессирующим течением заболевания ребенок консультирован врачом-генетиком. Рост 68 см, масса тела 6,3 кг. Осмысленного контакта нет. Взгляд в глаза не фиксирует. Нецеленаправленная двигательная активность симметричная. В руках игрушки не удерживает. На голос матери не реагирует. Микробрахицефалия. Вертикальный рост волос. Волосы густые. Гипертрихоз. Узкий лоб. Короткие глазные щели с длинными прямыми ресницами. Гипоплазия орбит. Эпикант. Альтернирующий страбизм. Короткий нос. Короткий фильтр. Диспластичные ушные раковины. Гипотония. Голову не удерживает, в положении на животе не приподнимает. Мраморный рисунок на коже. Клинодактилия мизинцев кистей. Крипторхизм. Гипоплазия мошонки. Кариотип 47,ХХY. Учитывая резистентность к терапии антиэпилептическими препаратами, проведено исследование гена SCN1A – патогенных вариантов не обнаружено.
Данные МРТ
По результатам магнитно-резонансной томографии (МРТ) с индукцией магнитного поля 3 Тл со сверхпроводящим магнитом MEXL-3010/G5 (Toshiba, Япония) выявлено расширение субарахноидальных пространств и желудочков мозга.
Данные видео-ЭЭГ-мониторинга с записью сна
Видео-ЭЭГ-мониторинг выполнен на модульной нейродиагностической системе NicoletOne (VIASYS Healthcare, США). Выявлены диффузные изменения биоэлектрической активности головного мозга. Замедление фоновой ритмики: в фоне доминирует диффузная тета-, дельта-активность с преобладанием в задних отделах полушарий амплитудой до 130 мкВ. Основной ритм и другие физиологические паттерны бодрствования во время исследования не зарегистрированы. При проведении функциональных проб значимого изменения фоновой ритмики не отмечено.
Сон дифференцирован на стадии, физиологические паттерны сна выражены удовлетворительно, представлены веретенами сна и К-комплексами (в отдельных случаях «эпилептизированы»). На этом фоне регистрируется эпилептиформная активность, представленная комплексами «острая – медленная волна», «пик-, полипик – медленная волна» амплитудой 100–200 мкВ, в виде региональных разрядов в левой и правой затылочных, затылочно-височно-теменных областях, независимо, изредка с тенденцией к распространению.
Индекс эпилептиформной активности в бодрствовании варьируется в пределах низких и средних значений, во сне – в пределах средних и высоких значений. На отдельных эпохах сна с высоким индексом эпилептиформных разрядов формируется картина модифицированной (заднепроекционной) гипсаритмии. После пробуждения зарегистрирована серия субклинических ЭЭГ-паттернов, характерных для тонических эпилептических спазмов, в виде появления коротких пробегов заднепроекционной быстроволновой ритмичной активности (англ. fast activity) амплитудой до 60 мкВ, длительностью 1–1,5 с. Клинически выраженных эпилептических приступов во время исследования не зарегистрировано (рис. 1).
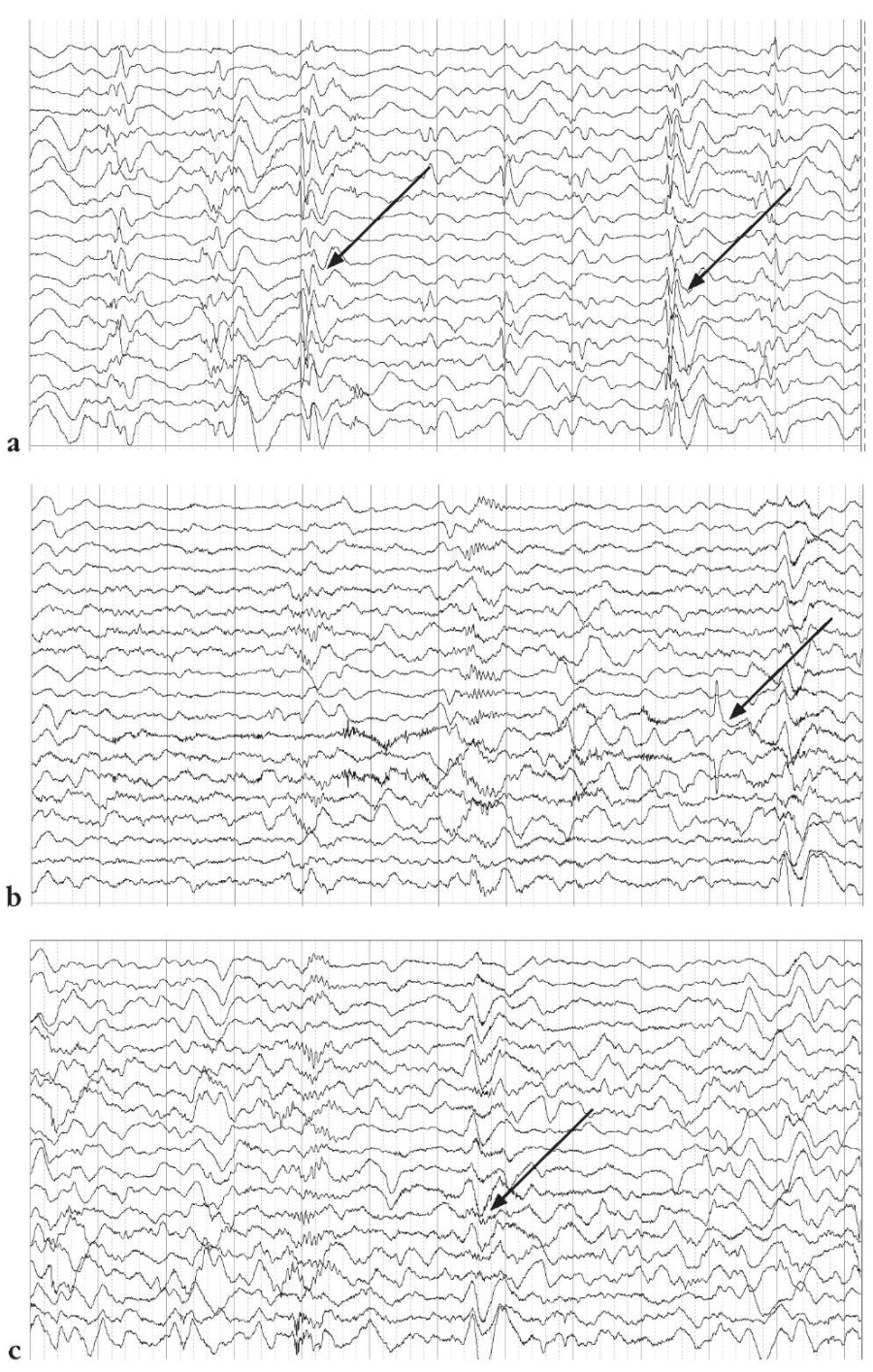
Рисунок 1. Результаты электроэнцефалографии (ЭЭГ) от 19.11.2022 г.:
стрелками указаны субклинические ЭЭГ-паттерны,
характерные для тонических эпилептических спазмов,
в виде появления коротких пробегов заднепроекционной быстроволновой
ритмичной активности амплитудой до 60 мкВ, длительностью 1–1,5 с (а–с)
Figure 1. Electroencephalography (EEG) imaging dated 19.11.2022:
arrows depict subclinical EEG patterns typical to tonic epileptic spasms
observed as emerging short runs of posterior projection fast activity
with an amplitude of up to 60 μV, duration 1–1.5 sec (a–c)
При повторной госпитализации в ГБУЗ «НПЦ спец. мед. помощи детям ДЗМ» (05–25.11.2022 г.) в терапию введен вигабатрин, поскольку приступы продолжались и при обследовании по данным ЭЭГ выявлена гипсаритмия, серия субклинических ЭЭГ-паттернов спазмов. На фоне применения вальпроевой кислоты (210 мг/сут) и вигабатрина (625 мг/сут), а также возникновения пневмонии серийные спазмы периодически повторялись.
С ноября 2022 г. получал комбинацию «вигабатрин (1500 мг/сут) + вальпроат натрия (250 мг/сут) + топирамат (62,5 мг/сут)» с положительной динамикой. Последний приступ в виде задержки дыхания, цианоза лица в течение 1 мин отмечен 06.02.2023 г. Серийные спазмы клинически не регистрировались.
Ребенок часто болел с рождения, периодически (1 раз в 1–3 мес) госпитализировался по поводу бронхитов и пневмоний. Вероятно, имел место микроаспирационный синдром.
В возрасте 1 год 4 мес (01.04.2023 г.) по экстренным показаниям (подъем температуры до 37,7 °С, появились кашель, дистантные хрипы, участились судорожные приступы) доставлен в стационар ДГКБ № 9. В связи с ухудшением состояния (очаговая неврологическая симптоматика, выраженная дыхательная недостаточность) ребенок переведен в ОРИТ ГБУЗ «НПЦ спец. мед. помощи детям ДЗМ» с целью коррекции антиконвульсантной терапии и респираторной реабилитации.
Неврологический статус
Общее состояние тяжелое за счет дыхательной недостаточности 2-й степени, метаболических нарушений, симптомов интоксикации, продолжающихся миоклоний как эпилептического, так и неэпилептического генеза. Сознание: поверхностное оглушение. Положение вынужденное. Питание пониженное. По шкале комы Глазго 13 баллов, по педиатрической шкале последовательной органной дисфункции (англ. рediatric Sequential Organ Failure Assessment, pSOFA) 4 балла. Общемозговой, менингеальной симптоматики нет. Судорог на момент осмотра нет. Зрачки: D=S. Фотореакция живая. Корнеальные рефлексы сохранены. OU – периодическое слабо выраженное расходящееся косоглазие. Нистагма на момент осмотра не выявлено. Глоточные, нёбные рефлексы снижены. Гиперкинезы генерализованные. Целенаправленных движений нет, диффузная мышечная гипотония, сухожильные рефлексы снижены, D=S. Выраженная задержка психомоторного развития: не держит голову, не переворачивается, взгляд фиксирует кратковременно, не прослеживает.
Дыхание
Фракция кислорода во вдыхаемой смеси (FIO2) – 31%, уровень насыщения крови кислородом (SPO2) – 97%. Дотация O2, способ дотации – лицевая маска, поток O2 – 2 л/мин. Частота дыхательных движений – 30 в минуту. Дыхание справа жесткое, ослаблено в средних отделах, слева ослаблено по всем полям, значительно хуже проводится в базальных и латеральных отделах. По всем полям влажные мелкопузырчатые хрипы, больше слева, непостоянная крепитация. При дыхании воздухом снижение сатурации до 80%, нарастание одышки, акроцианоз.
Состояние сердечно-сосудистой системы
Гемодинамика стабильная. Систолическое давление – 87 мм рт. ст., диастолическое давление – 47 мм рт. ст., среднее артериальное давление – 60 мм рт. ст. Частота сердечных сокращений (ЧСС) – 137 уд/мин, ритм сердца не нарушен. Тоны сердца приглушенные. Rg-картина соответствует двусторонним полисегментарным инфильтративным изменениям, без отрицательной динамики. По сравнению с данными компьютерной томографии от 17.04.2023 г. динамика отрицательная в виде увеличения объема перибронхиальной инфильтрации.
МРТ-картина
МРТ-картина от 20.04.2023 г. с отрицательной динамикой: признаки дисмиелинизации белого вещества больших полушарий, гемисфер мозжечка и ствола мозга, патологические изменения в подкорковых структурах. Наружно-внутренняя сообщающаяся гидроцефалия, увеличение размеров желудочковой системы и цистернальных пространств. Признаки двустороннего среднего отита и пансинусита (рис. 2).
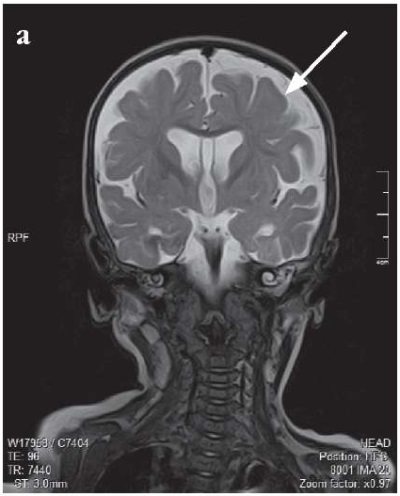
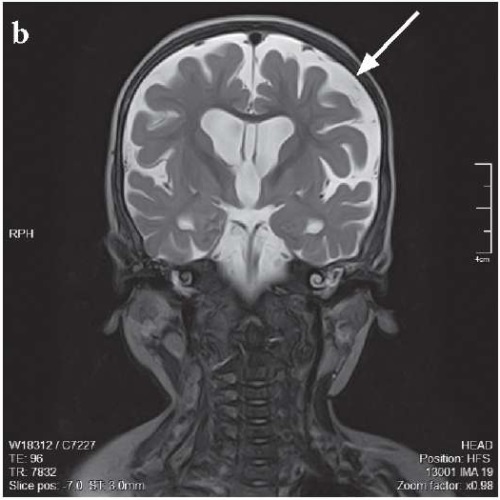
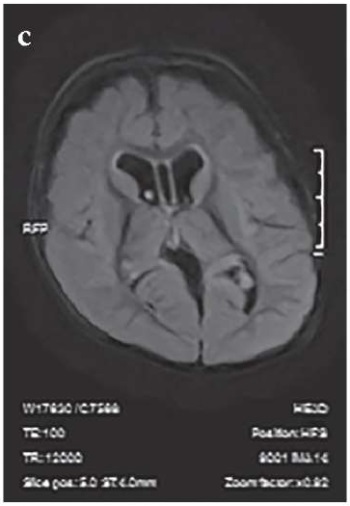
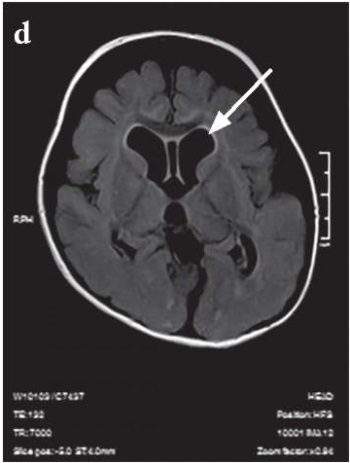
Рисунок 2. Результаты магнитно-резонансной томографии головного мозга
от 11.10.2022 г. (a, c) и 20.04.2023 г. (b, d).
Стрелками указаны прогрессирующая дисмиелинизация белого вещества больших полушарий,
гемисфер мозжечка и ствола мозга, патологические изменения в подкорковых структурах (a, b)
и наружно-внутренняя сообщающаяся гидроцефалия,
увеличение размеров желудочковой системы и цистернальных пространств (d)
Figure 2. Brain magnetic resonance imaging dated 11.10.2022 (a, c) and 20.04.2023 (b, d).
Arrows depict progressive white matter dysmyelination in the cerebral hemispheres,
cerebellar hemispheres and brainstem, pathological changes in the subcortical structures (a, b)
and external-internal communicating hydrocephalus,
enlarged ventricular system and cisternal spaces (d)
Ухудшение состояния
20.04.2023 г. в 11 ч 00 мин у ребенка появились выраженные клонии мышц грудной клетки, сопровождающееся ригидностью грудной клетки, вследствие чего развился бронхоспазм со снижением сатурации до 52%, брадикардией до 45 уд/мин. Проводилась противосудорожная, бронхолитическая, противоотечная, инотропная терапия, на этом фоне клонии и бронхоспазм были купированы, сатурация 97–98%, аускультативно дыхание проводилось во все отделы, ребенок был реинтубирован оротрахеально, на фоне введения атропина и допамина (5 мкг/кг/мин) гемодинамика стабильная: артериальное давление 105/65 мм рт. ст, ЧСС 140–150 уд/мин, ритм синусовый. Через 2 ч инотропная поддержка была отменена.
Выполнена рентгенография органов грудной клетки: картина соответствовала перибронхиальной инфильтрации. По кислотно-щелочному состоянию – респираторный ацидоз, гиперкарбия. По данным электрокардиографии: умеренная синусовая брадикардия 107 уд/мин, электрическая ось сердца в горизонтальном положении. Замедление проводимости по правой ножке пучка Гиса. Элевация сегмента ST в отведениях II, III, AVF, V5, V6 до 0,5 мм. Интервал P–Q на нижней границе нормы. По результатам эхокардиографии: структуры сердца сформированы правильно, полости сердца не расширены, сократительная способность миокарда в пределах нормы. Диурез умеренно снижен (1,6 мл/кг/ч).
В дальнейшем состояние ребенка прогрессивно ухудшалось. В связи с наличием дисфагии на фоне неврологических нарушений и аспирационного синдрома, частыми пневмониями с целью разобщения дыхательного и пищеварительного трактов была проведена гастростомия с применением видеоэндоскопической техники.
11.05.2023 г. у ребенка с тяжелой сочетанной патологией на фоне проводимого лечения ввиду активации инфекционного процесса, отрицательной соматической динамики с возникновением полиорганной недостаточности развился летальный исход. Механизм смерти: сердечно-сосудистая и дыхательная недостаточность.
Полноэкзомное секвенирование и секвенирование по Сэнгеру выполнено в генетической лаборатории ГБУЗ «НПЦ спец. мед. помощи детям ДЗМ» и ООО «Геноаналитика». Геномная ДНК выделена методом лизиса клеток с последующей очисткой на стекловолоконных фильтрах (реактивы QIAamp DNA Mini Kit, Qiagen), затем использована для приготовления геномных библиотек для массового параллельного секвенирования (NEBNext Ultra II, New England BioLabs). Из полученных библиотек методом гибридизации были отобраны только те участки ДНК, которые соответствуют экзонам генов и сайтам сплайсинга (SureSelect AllExon V7, Agilent). Далее было проведено определение их нуклеотидной последовательности на секвенаторе HiSeq 1500 (Illumina, США), реактивы HiSeq Rapid SBS Kit v2.
Результаты полноэкзомного секвенирования получены после наступления летального исхода. Выявлен ранее не описанный вариант нуклеотидной последовательности в 17-м экзоне гена SEMA6B в гетерозиготном состоянии, приводящий к сдвигу рамки считывания начиная с 836-й позиции белка.
Мутации в гене SEMA6B в гетерозиготном состоянии описаны у пациентов с ПМЭ, что предполагает аутосомно-доминантный тип наследования. Частота выявленного варианта нуклеотидной последовательности в контрольной выборке gnomAD в гетерозиготном состоянии составляет 0,0006%, в европейской популяции – 0%. Выявленный вариант нуклеотидной последовательности не зарегистрирован в контрольной выборке RUSeq. Алгоритмы предсказания патогенности в данном случае неприменимы. Поскольку выявленный вариант нарушает синтез полнофункционального белка, его следует расценивать как вероятно патогенный вариант согласно критериям Американской коллегии медицинской генетики и геномики (англ. American College of Medical Genetics and Genomics, ACMG).
Обнаруженный вариант нуклеотидной последовательности был валидирован методом секвенирования по Сэнгеру. В связи со смертью ребенка согласия на исследование происхождения варианта в гене не получено.
Ген SEMA6B локализован на хромосоме 19p13 и включает 17 кодирующих экзонов и сайт связывания PPAR [9]. Благодаря использованию иммуноокрашивания К. Hamanaka et al. определили экспрессию гена SEMA6B в нейронах в нескольких областях мозга, включая кору головного мозга, мозжечковые клетки Пуркинье и интернейроны; определенные типы клеток включали возбуждающие и ГАМК-ергические1 тормозные нейроны [3]. SEMA6B является представителем семейства белков семафоринов класса 6, которые участвуют в различных аспектах развития нервной системы, включая миграцию клеток нервного гребня, развитие аксонов и мозжечка [3][4]. Впервые К. Hamanaka et al. в 2020 г. установили связи вариантов в гене SEMA6B с развитием ПМЭ [3].
Варианты в гене SEMA6B связаны с широким фенотипическим спектром – от энцефалопатии развития без эпилепсии до эпилептической энцефалопатии. Однако относительно частым проявлением является ПМЭ – фенотип, ранее определенный Марсельским консенсусом. ПМЭ характеризуется задержкой развития, прогрессирующей умеренной или тяжелой умственной отсталостью и моторной регрессией вследствие ухудшения миоклонуса и дополнительного неврологического дефицита. У описанного нами пациента наблюдались тяжелые прогрессирующие неврологические и когнитивные нарушения [10].
Согласно базам данных к настоящему времени описано 29 пациентов с 22 различными вариантами нуклеотидной последовательности в гене SEMA6B, 18 из которых локализованы в 17-м экзоне гена. Варианты в гене SEMA6B включают 5 миссенс-вариантов, 5 нонсенс-вариантов и 12 вариантов со сдвигом рамки считывания (рис. 3) [1].
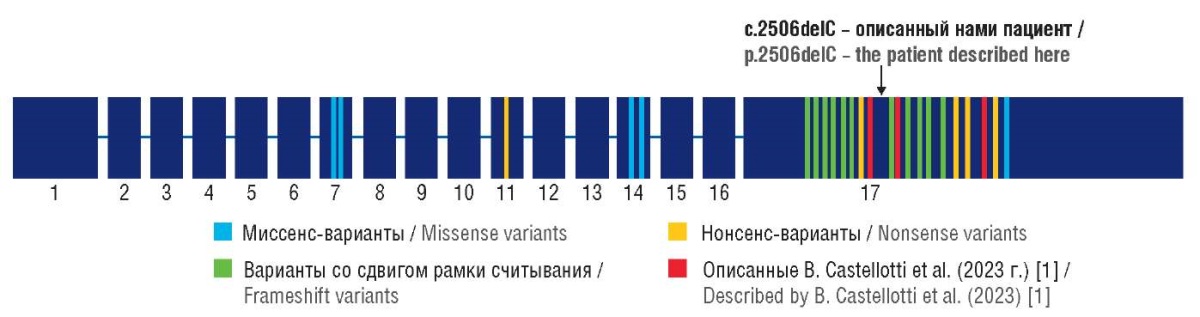
Рисунок 3. Структура гена SEMA6B
и описанные варианты нуклеотидной последовательности [1]
Figure 3. SEMA6B gene structure and described nucleotide sequence variants [1]
Для обзора литературы и сравнения с клиническим проявлением заболевания у нашего пациента мы использовали базу данных PubMed/MEDLINE и учитывали следующую информацию: тип варианта, экзон, ранний(е) признак(и) заболевания, особенности фенотипа, тип судорог, ответ на терапию, наличие когнитивной регрессии, атаксия, спастичность / пирамидные симптомы, миоклонус, данные ЭЭГ и МРТ головного мозга [1][5–8].
Судороги были у 25 из 29 пациентов, в т.ч. у 2 больных с бессудорожным эпилептическим статусом. Возраст начала заболевания варьировался от 11 мес до 10 лет. У 16 пациентов наблюдалось прогрессирующее нарушение моторики. В 10 случаях сообщалось об атаксии и спастичности. У 10 человек отмечена тяжелая когнитивная регрессия с потерей речи. У 16 больных были миоклонические подергивания, из них только 10 имели четкий фенотип ПМЭ с выраженным миоклонусом. В остальных 6 случаях наблюдались миоклонические подергивания на фоне полиморфных припадков, предполагалась энцефалопатия развития и эпилептическая энцефалопатия (синдром Леннокса–Гасто или эпилепсия с миоклоническими атоническими судорогами). У 3 пациентов описано более легкое течение заболевания, в 1 случае состояние характеризовалось только наличием эпилепсии.
На рисунке 4 показаны генотип-фенотипические корреляции у пациентов с вариантами в гене SEMA6B [1].
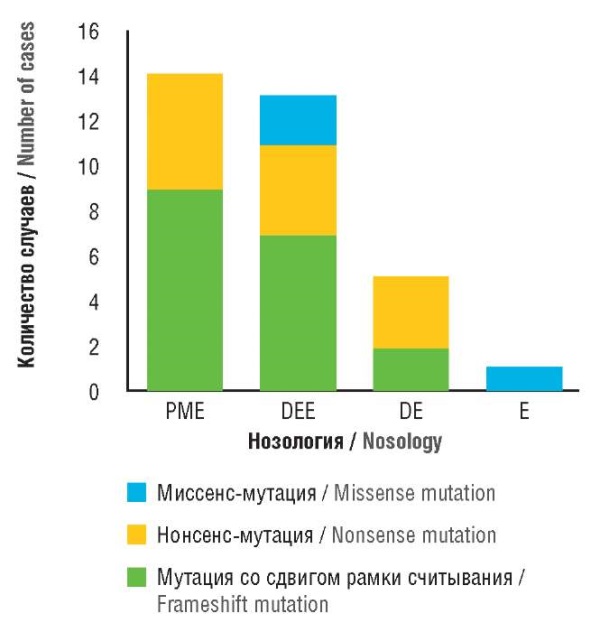
Рисунок 4. Генотип-фенотипические корреляции у пациентов с вариантами в гене SEMA6B [1].
PME (англ. progressive myoclonus epilepsy) –
прогрессирующая миоклонус-эпилепсия;
DEE (англ. developmental and epileptic encephalopathy) –
энцефалопатия развития и эпилептическая энцефалопатия;
DE (англ. developmental encephalopathy) – энцефалопатия развития;
E (англ. epilepsy) – эпилепсия
Figure 4. Genotype-phenotype correlations in patients with SEMA6B gene variants [1].
PME – progressive myoclonus epilepsy;
DEE – developmental and epileptic encephalopathy;
DE – developmental encephalopathy; E – epilepsy
Варианты со сдвигом рамки считывания наиболее часто связаны с фенотипом ПМЭ. К. Hamanaka et al. предположили, что этот тип мутации может приводить к «ускользанию» от нонсенс-опосредованного распада мРНК. Таким образом, трансляция укороченных белков может оказывать токсическое действие или изменять передачу сигналов семафорина [3].
Как и в большинстве ранее зарегистрированных случаев, у нашего пациента был выявлен патогенный вариант в последнем, 17-м экзоне гена SEMA6B, приводящий к сдвигу рамки считывания и синтезу измененного укороченного белка. В клинической картине у больного с ранним началом заболевания в возрасте 8 мес мы наблюдали прогрессирующее тяжелое течение эпилепсии с последующим развитием эпилептического статуса и летальным исходом.
Молекулярная диагностика у пациентов с нарушениями развития нервной системы часто вызывает затруднения как из-за неспецифического фенотипа, так и в связи с генетической гетерогенностью. Представленный клинический случай прогрессирующей миоклонус-эпилепсии, обусловленной вариантом мутации в гене SEMA6B, у ребенка с синдромом Клайнфельтера расширил фенотипический и генетический спектр. При принятии решения о тактике обследования таких пациентов необходимо проведение генетического тестирования методом полноэкзомного или полногеномного секвенирования даже в случаях выявления хромосомной аномалии у пробанда.
1. ГАМК – гамма-аминомасляная кислота.
1. Castellotti B., Canafoglia L., Freri E., et al. Progressive myoclonus epilepsies due to SEMA6B mutations. New variants and appraisal of published phenotypes. Epilepsia Open. 2023; 8 (2): 645–50. https://doi.org/10.1002/epi4.12697.
2. Franceschetti S., Michelucci R., Canafoglia L., et al. Progressive myoclonic epilepsies: definitive and still undetermined causes. Neurology. 2014; 82 (5): 405–11. https://doi.org/10.1212/WNL.0000000000000077.
3. Hamanaka K., Imagawa E., Koshimizu E., et al. De novo truncating variants in the last exon of SEMA6B cause progressive myoclonic epilepsy. Am J Hum Genet. 2020; 106 (4): 549–58. https://doi.org/10.1016/j.ajhg.2020.02.011.
4. Yazdani U., Terman J.R. The semaphorins. Genome Biol. 2006; 7: 211. https://doi.org/10.1186/gb-2006-7-3-211.
5. Shu L., Xu Y., Tian Q., et al. A frameshift variant in the SEMA6B gene causes global developmental delay and febrile seizures. Neurosci Bull. 2021; 37 (9): 1357–60. https://doi.org/10.1007/s12264-021-00717-5.
6. Xiaozhen S., Fan Y., Fang Y., et al. Novel truncating and missense variants in SEMA6B in patients with early-onset epilepsy. Front Cell Dev Biol. 2021; 9: 633819. https://doi.org/10.3389/fcell.2021.633819.
7. Duan J., Chen Y., Hu Z., et al. Non-convulsive status epilepticus in SEMA6B-related progressive myoclonic epilepsy: a case report with literature review. Front Pediatr. 2022; 10: 859183. https://doi.org/10.3389/fped.2022.859183.
8. Cordovado A., Schaettin M., Jeanne M., et al. SEMA6B variants cause intellectual disability and alter dendritic spine density and axon guidance. Hum Mol Genet. 2022; 31 (19): 3325–40. https://doi.org/10.1093/hmg/ddac114.
9. Correa R.G., Sasahara R.M., Bengtson M.H., et al. Human semaphoring 6B [(HSA)SEMA6B], a novel human class 6 semaphorin gene: alternative splicing and all-trans-retinoic aciddependent downregulation in glioblastoma cell lines. Genomics. 2001; 73 (3): 343–8. https://doi.org/10.1006/geno.2001.6525.
10. Classification of progressive myoclonus epilepsies and related disorders. Marseille Consensus Group. Ann Neurol. 1990; 28 (1): 113–6. https://doi.org/10.1002/ana.410280129.
Кожанова Татьяна Викторовна – к.м.н., ведущий научный сотрудник, врач – лабораторный генетик; доцент кафедры неврологии, нейрохирургии и медицинской генетики им. академика Л.О. Бадаляна педиатрического факультета
ул. Авиаторов, д. 38, Москва 119620, Россия
ул. Островитянова, д. 1, Москва 117997, Россия
Жилина Светлана Сергеевна – к.м.н., ведущий научный сотрудник, врач-генетик; доцент кафедры неврологии, нейрохирургии и медицинской генетики им. академика Л.О. Бадаляна педиатрического факультета
ул. Авиаторов, д. 38, Москва 119620, Россия
ул. Островитянова, д. 1, Москва 117997, Россия
Мещерякова Татьяна Ивановна – к.м.н., ведущий научный сотрудник, врач-генетик; доцент кафедры общей и медицинской генетики медико-биологического факультета
ул. Авиаторов, д. 38, Москва 119620, Россия
ул. Островитянова, д. 1, Москва 117997, Россия
Сушко Лилия Марленовна – врач-невролог
ул. Авиаторов, д. 38, Москва 119620, Россия
Осипова Карина Вартановна – к.м.н., заведующая психоневрологическим отделением № 1
ул. Авиаторов, д. 38, Москва 119620, Россия
Мазур Александр Михайлович – к.ф.-м.н., директор по науке
ул. Ленинские горы, вл. 1, стр. 77, Москва 119234, Россия
Фоменко Сергей Сергеевич – биоинформатик
ул. Ленинские горы, вл. 1, стр. 77, Москва 119234, Россия
Крапивкин Алексей Игоревич – д.м.н., директор
ул. Авиаторов, д. 38, Москва 119620, Россия
Заваденко Николай Николаевич – д.м.н., профессор, заведующий кафедрой неврологии, нейрохирургии и медицинской генетики им. академика Л.О. Бадаляна педиатрического факультета
ул. Островитянова, д. 1, Москва 117997, Россия
Кожанова Т.В., Жилина С.С., Мещерякова Т.И., Сушко Л.М., Осипова К.В., Мазур А.М., Фоменко С.С., Крапивкин А.И., Заваденко Н.Н. SEMA6B-ассоциированная прогрессирующая миоклонус-эпилепсия у пациента с синдромом Клайнфельтера. Эпилепсия и пароксизмальные состояния. 2024;16(1):45-53. https://doi.org/10.17749/2077-8333/epi.par.con.2024.175
Kozhanova T.V., Zhilina S.S., Meshcheryakova T.I., Sushko L.M., Osipova K.V., Mazur A.M., Fomenko S.S., Krapivkin A.I., Zavadenko N.N. SEMA6B-related progressive myoclonus epilepsy in a patient with Klinefelter syndrome. Epilepsy and paroxysmal conditions. 2024;16(1):45-53. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2024.175

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































