



Содержание
Перейти к:
https://doi.org/10.17749/2077-8333/epi.par.con.2024.182
Перейти к:
Болезнь де Виво является редким генетическим нарушением, связанным с дефицитом транспортера глюкозы 1-го типа (англ. glucose transporter type 1, GLUT1). В статье представлен обзор публикаций, в которых описаны различные клинические проявления заболевания, включая сочетание эпилепсии с хореическим гиперкинезом. Отмечается резистентность приступов к базовой антиэпилептической терапии, в качестве основного метода лечения предлагается кетогенная диета. Приведено собственное клиническое наблюдение 18-летнего пациента, у которого с 1,5 лет впервые возникли миоклонико-астатические приступы и атактические проявления в виде нарушения координации движений и неустойчивости при ходьбе. На фоне терапии препаратами вальпроевой кислоты приступы сохранялись с частотой до 5 раз в месяц. С 17 лет появились непроизвольные насильственные нерегулярные движения мышц туловища и конечностей, совершающиеся в быстром темпе. Проведено комплексное обследование, в результате верифицирован диагноз «болезнь де Виво». Путем назначения адекватной противоэпилептической терапии и кетогенной диеты удалось стабилизировать состояние больного, купировать проявления эпилепсии и гиперкинетического расстройства. Мы обращаем внимание специалистов на дифференциальную диагностику состояний, характеризующихся эпилептическими приступами, умственной отсталостью и насильственными движениями, а также на диагностику и тактику ведения пациентов с болезнью де Виво. К сожалению, не все больные с данной патологией получают адекватную патогенетическую и симптоматическую терапию. Зачастую пациенты подвергаются многочисленным госпитализациям, т.к. не диагностирована основная причина симптомов, а именно дефицит GLUT1.
Шова Н.И., Михайлов В.А., Романюго Г.Д. Болезнь де Виво (сочетание миоклонико-астатической эпилепсии и хореи): литературный обзор, описание клинического наблюдения. Эпилепсия и пароксизмальные состояния. 2024;16(2):145-156. https://doi.org/10.17749/2077-8333/epi.par.con.2024.182
Shova N.I., Mikhailov V.A., Romanyugo G.D. De Vivo disease (myoclonic-astatic epilepsy combined with chorea): literature review, clinical case description. Epilepsy and paroxysmal conditions. 2024;16(2):145-156. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2024.182
Недостаток глюкозы в головном мозге (нейрогликопения) в ранние постнатальные годы, вероятно, останавливает развитие нейронных цепей, лежащих в основе разнообразного репертуара скоординированных движений и сложного поведения. Один из способов начать понимать клеточную патологию и молекулярные механизмы, связанные с нейрогликопенией, – это изучение моногенных причин состояния. Типичным примером является синдром дефицита транспортера глюкозы 1-го типа (англ. glucose transporter type 1 deficiency syndrome, GLUT1DS), или болезнь де Виво (OMIM #606777, ORPHA71277).
С момента первоначального описания GLUT1DS в 1991 г. [1] число пациентов с данным заболеванием неуклонно росло, чему способствовало появление молекулярной диагностики. В недавно опубликованном шотландском проспективном популяционном исследовании сообщалось о частоте рождений 1 случай на 24 тыс. случаев эпилепсии в первые три года [2]. В соответствии с прогнозируемой заболеваемостью 1,65–2,22 случая на 100 тыс. рождений [3] это, вероятно, минимальная частота, поскольку GLUT1DS может проявляться позже эпилепсией или двигательными расстройствами. В ретроспективных исследованиях распространенность GLUT1DS оценивалась в 1 случай на 83 тыс. в Дании [4] и 1 случай на 90 тыс. в Австралии [5]. Поражаются все возрастные группы, от младенцев до взрослых.
В 1998 г. было обнаружено, что гаплонедостаточность гена SLC2A1 и, следовательно, низкие уровни его транслируемого продукта, белка GLUT1, лежат в основе GLUT1DS [6]. Ген SLC2A1 состоит из 10 экзонов и 9 интронов, локализуется на коротком плече хромосомы 1. Описано более 150 мутаций в данном гене. Большинство пациентов имеют мутации de novo и также могут наследовать заболевание по аутосомно-доминантному типу [7]. В редких случаях наблюдается аутосомно-рецессивный тип наследования GLUT1DS, что может привести к сложным гетерозиготам [8].
Выявление генетической причины GLUT1DS способствовало точной диагностике заболевания. Это также привело к распознаванию значительно расширенного клинического фенотипа. Существует разнообразный спектр клинических проявлений, начиная от наблюдаемых при классической форме расстройства и заканчивая такими особенностями, как некинезигенные дискинезии, спастические параплегии и гемолитические анемии [9][10].
Хотя принято считать, что настоящий случай GLUT1DS связан с молекулярными повреждениями в гене SLC2A1, мутации в нем не являются существенными для запуска клинического фенотипа, соответствующего дефициту GLUT1 [11]. В некоторых случаях у подобных пациентов обнаруживается пониженная экспрессия транспортера, несмотря на нормальную последовательность, кодирующую белок [12]. Это наводит на мысль о некодирующей мутации в гене SLC2A1 или нарушении в факторах, которые регулируют экспрессию GLUT1. Также возможно, что новые механизмы, воздействующие скорее на активность, чем на ее экспрессию, объясняют гипогликорахию и клинический фенотип.
Синдром может быть представлен двумя клиническими формами:
Клинические симптомы провоцируются физической нагрузкой, голодом с некоторым улучшением состояния после еды.
Фармакорезистентные эпилептические приступы часто являются первым признаком заболевания. Могут наблюдаться любые, различные по своей семиотике типы припадков [13]. При этом генерализованные формы встречаются чаще, чем фокальные [14, 15]. Абсансная эпилепсия с ранним началом (в возрасте до 4 лет) и эпилепсия с миоклонически-астатическими припадками (синдром Дузе) были связаны с патогенными вариантами в гене SLC2A1 [16].
Вторым по значимости признаком являются непроизвольные кратковременные движения головы по типу кивков с повторяющимися разнонаправленными движениями глаз [17]. Позже в детстве возникают другие пароксизмальные явления, но их тяжесть сильно варьируется (возможны гиперкинезы, парезы/плегии, атаксии). По мере взросления пациента происходят постепенное улучшение клинической картины, снижение частоты и тяжести пароксизмальных явлений. Пароксизмальные немоторные эпизоды включают мигрень, поведенческие нарушения, циклическую рвоту и диссомнические расстройства [18].
Классически данные нарушения присутствуют до приема пищи и смягчаются ее приемом. Стойкие двигательные расстройства представлены спастичностью, атаксией и дистонией, часто приводящими к нарушениям походки с последующей хореей и тремором [19]. Атаксия становится все более очевидной в позднем младенчестве, когда ребенок стоит и начинает ходить. Атаксия больше туловищная. Хорея часто бывает легкой и поражает лицо и дистальные отделы верхних конечностей. Терминальный интенционный тремор встречается часто и обычно связан с другими признаками дисфункции мозжечка. Миоклонус, как правило, эпилептический, неэпилептический миоклонус наблюдается реже и включает миоклонус вздрагивания, миоклонус действия и постуральный миоклонус. Диспраксия недостаточно распознана, она бывает глазодвигательной и оробуккальной. Пароксизмальные двигательные расстройства отмечаются примерно у 75% пациентов. Потенциальными триггерами являются эмоциональный стресс, лихорадка, усталость, недостаточный кетоз, лишение сна, перепады температуры и прием лекарств.
Атипичные проявления включают писчий спазм, перемежающуюся атаксию, тетраплегию, синдром беспокойных ног, крампи-синдром. У отдельных пациентов описаны перемежающаяся детская гемиплегия, гемиплегическая мигрень, циклическая рвота и инсультоподобные эпизоды с пароксизмальным гемипарезом, дизартрией или афазией [20].
У больных с GLUT1DS когнитивный дефицит сильно варьируется. Часто наблюдаются задержка речи и трудности с экспрессивной речью (возможно, связанные с нарушениями речи, такими как дизартрия), трудности в обучении и когнитивные расстройства. Последние могут быть легкими, среднетяжелыми или тяжелыми, но без определенного нейропсихологического профиля [21–24]. Когнитивные нарушения обычно пропорциональны возрасту начала и тяжести неврологических проявлений [22].
GLUT1DS связан с умственной отсталостью от легкой до тяжелой степени, причем степень тяжести пропорциональна общей тяжести заболевания. Дизартрия с различной степенью нарушения речи отмечается у всех больных [25]. Также могут наблюдаться нарушения поведения, синдром дефицита внимания и гиперактивности и депрессия [25][26].
Секвенирование гена SLC2A1 по Сэнгеру является молекулярным методом выбора для характеристики GLUT1DS. Должны быть секвенированы все 10 экзонов, включая по меньшей мере 10 специфических фланкирующих интронных оснований каждого из них. Если секвенирование отрицательное, следует выявлять грубые делеции или дупликации с использованием методологии, чувствительной к этому типу вариантов, такой как мультиплексная лигазозависимая амплификация зонда (англ. multiplex ligation-dependent probe amplification, MLPA).
Массовое параллельное секвенирование и сравнительная геномная гибридизация дают преимущество изучения гена SLC2A1 вместе с большим количеством генов, что может быть полезно для целей дифференциальной диагностики [27].
После того как дефект гена SLC2A1 охарактеризован и подтвержден молекулярно, рекомендуется провести генетические исследования у родителей пациента, чтобы исключить семейный источник заболевания [28].
Лишь у небольшой доли больных с клиническим диагнозом GLUT1DS молекулярное исследование гена SLC2A1 будет отрицательным [29]. Возможно, в этих случаях патогенный вариант локализован в областях гена, которые обычно не изучаются (промоторные и глубокие интронные области) [29–31]. Недавно было высказано предположение, что патогенные варианты в других генах могут вызывать симптомы, подобные GLUT1DS, либо сами по себе, либо косвенно, нарушая функцию гена SLC2A1 [32][33].
Разработан новый метод идентификации с использованием секвенирования всего экзома, который включает обнаружение редких гомозиготных миссенс-вариантов (c.526C>T (p.Arg176Trp) и c.629C>T (p.Ala210Val)) в гене SLC45A1, кодирующих другой церебральный транспортер глюкозы, что указывает на рецессивную мутацию в SLC45A1 (второй церебральный транспортер глюкозы в дополнение к GLUT1), которая вызывает умственную отсталость и эпилепсию. У лиц, протестированных в этой когорте, отсутствовали другие клинические особенности, связанные с GLUT1, а также не была зарегистрирована гипогликорахия.
Интериктальная электроэнцефалографическая (ЭЭГ) картина обычно нормальная. Однако в зависимости от возраста могут наблюдаться различные паттерны: у младенцев чаще наблюдается замедление и фокальная эпилептиформная активность, в то время как у детей в возрасте 2 лет и старше – генерализованная пиковая волна частотой 2,5–4 Гц. Интересной особенностью, если она присутствует, является аномальная ЭЭГ перед приемом пищи, которая улучшается при приеме пищи [34].
Пониженное содержание глюкозы в ликворе в сочетании с нормальным уровнем глюкозы в плазме крови – отличительный маркер для GLUT1DS [35][36]. Примерно у 90% пациентов уровень глюкозы ниже 2,2 ммоль/л [37]. Люмбальную пункцию следует проводить после 4–6 ч голодания, чтобы стабилизировать концентрацию глюкозы в спинномозговой жидкости [38]. Уровень глюкозы в крови следует измерять непосредственно перед люмбальной пункцией, т.к. это позволит избежать погрешности во время гипергликемии, которая может быть связана со стрессовой реакцией на данную манипуляцию.
У пациентов с GLUT1DS наблюдается пониженное содержание глюкозы в ликворе с нормальным или низким уровнем молочной кислоты, что помогает отличить его от других состояний, которые также связаны с гипогликорахией, таких как бактериальные инфекции центральной нервной системы и некоторые митохондриальные заболевания [39].
При проведении магнитно-резонансной томографии у пациентов с GLUT1DS отмечаются либо отсутствие характерных изменений [24], либо незначительные неспецифические отклонения, такие как легкая кортикальная гипотрофия [40] или нарушение миелинизации [41]. Картина аномалий развития белого вещества, включающая высокий T2-сигнал подкорковых U-волокон, описана у некоторых больных [35] – этот признак, вероятно, указывает на нарушение процесса миелинизации.
На основании вышеуказанных методов диагностики недостаточности GLUT1 в организме предлагается тактика ведения пациентов с подозрением на болезнь де Виво, представленная на рисунке 1 [42].
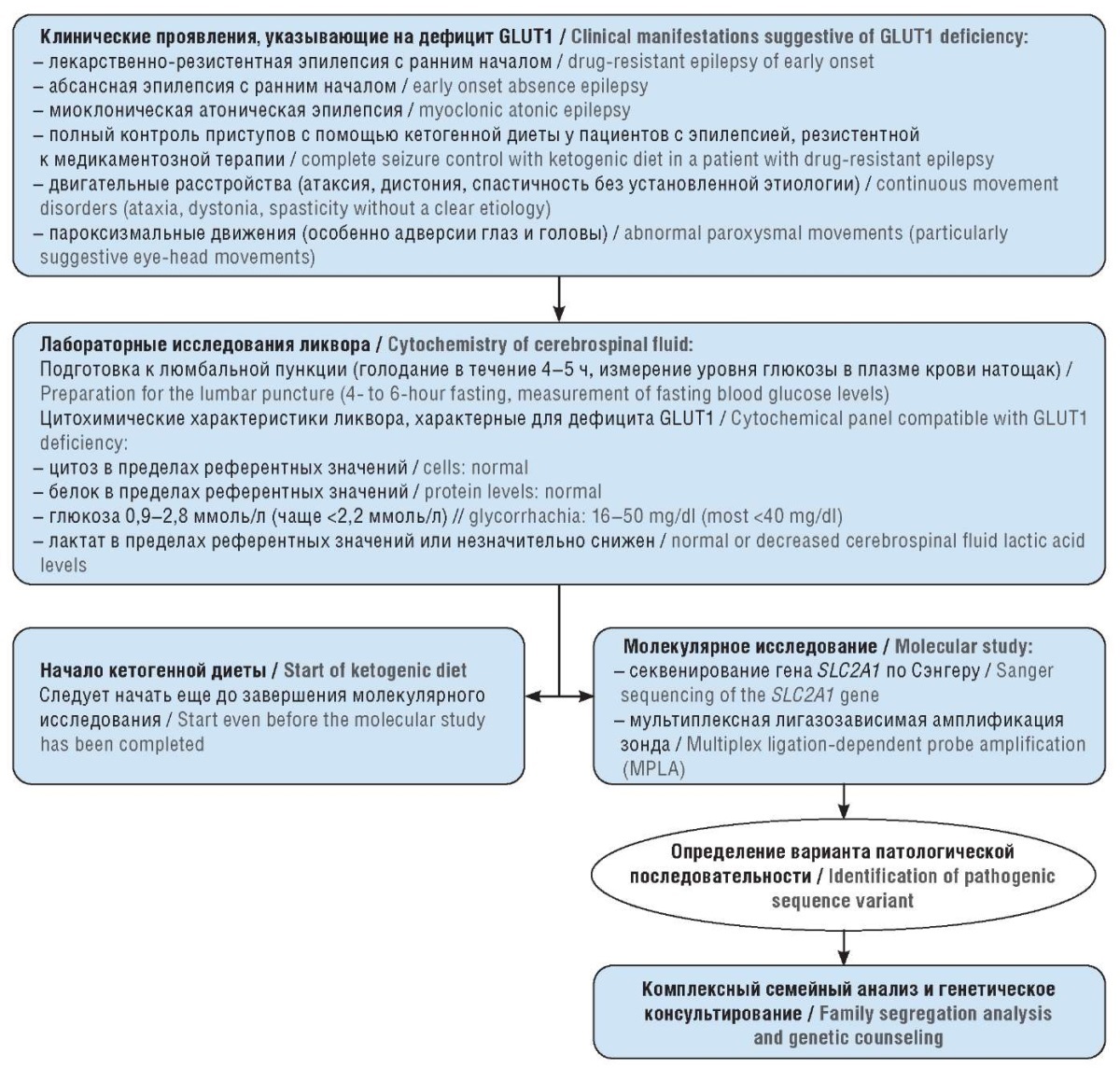
Рисунок 1. Алгоритм диагностики пациентов с дефицитом транспортера глюкозы 1-го типа
(англ. glucose transporter type 1, GLUT1) (адаптировано по [42])
Figure 1. The diagnostic algorithm for patients with glucose transporter type 1 (GLUT1) deficiency
(adapted from [42])
Таким образом, возникает необходимость проведения дифференциальной диагностики с заболеваниями, сопровождающимися нарушением метаболизма углеводов. Принципы дифференциальной диагностики указанных нозологий приведены в таблице 1.
Таблица 1. Дифференциальная диагностика заболеваний,
вызванных нарушением метаболизма углеводов
и сопровождающихся неврологическими расстройствами
Table 1. Differential diagnosis of diseases caused by impaired carbohydrate metabolism
and accompanied by neurological disorders
Параметр / Parameter | Болезнь де Виво / De Vivo disease | Синдром Ангельмана / Angelman syndrome | Семейный гиперинсулинизм / Familial hyperinsulinism | Синдром опсоклонус-миоклонус / Opsoclonus-myoclonus syndrome |
Специфический анамнез / Specific anamnesis | Врожденное заболевание с дебютом в младенчестве и раннем детстве / The congenital disease with onset in infancy and early childhood [2] | Врожденное заболевание с дебютом в 6–12 мес / The congenital disease with onset in the age of 6–12 months [43] | Врожденное заболевание с дебютом в неонатальном периоде / Congenital disease with onset in neonatal period [44] | Развивается после респираторной инфекции или является проявлением паранеопластического синдрома / Develops after respiratory infection or manifestates paraneoplastic syndrome [45] |
Неврологическая симптоматика / Neurological symptoms | Фармакорезистентная эпилепсия, задержка психического развития, дизартрия, двигательные расстройства / Pharmacoresistant epilepsy, mental retardation, dysarthria, motor disorders [42] | Выраженная задержка психического развития, атаксия и тремор конечностей, в большинстве случаев сочетающиеся с тонико-клоническими, миоклоническими или астатическими эпилептическими приступами / Severe mental retardation, ataxia and tremor of extremities, in most cases combined with tonic-clonic, myoclonic or astatic epileptic seizures [43] | Фармакорезистентная эпилепсия, астения, задержка психомоторного развития, нарушения зрения / Pharmacoresistant epilepsy, asthenia, delayed psychomotor development, visual impairment [44] | Вегетативные нарушения, сменяющиеся возникновением миоклонических подергиваний мышц лица и конечностей, опсоклонусом глазных яблок и мозжечковыми нарушениями / Vegetative disorders, followed by the appearance of myoclonic twitching of face and extremities muscles, ocular opsoclonus and cerebellar disorders [45] |
Анализ спинномозговой жидкости / Cerebrospinal fluid analysis | Гипогликорахия + гипо/нормолактатрахия // Hypoglycorrhachia + hypo/normolactatrhachia [42] | Без отклонений / No deviations [43] | Гипогликорахия / Hypoglicorrhachia [44] | Без отклонений / No deviations [46] |
Нейро-визуализация / Neuroimaging | Без отклонений либо незначительная гипотрофия головного мозга / No deviations, or minor brain hypotrophy [24] | Без отклонений либо умеренные атрофические изменения коры головного мозга, нейрональные гетеротопии или атрофия мозжечка / No deviations, either moderate atrophic changes in cerebral cortex, neuronal heterotopias or cerebellar atrophy [43] | Дистрофические и атрофические изменения преимущественно в затылочных отделах коры больших полушарий / Dystrophic and atrophic changes mainly in occipital cortex of cerebral hemispheres [47] | Без отклонений / No deviations [46] |
Специфическое лечение / Specific treatment | Кетогенная диета/ Ketogenic diet [48] | Вальпроевая кислота или бензодиазепины / Valproic acid or benzodiazepines [49] | Диазоксид и соблюдение высокоуглеводной диеты / Diazoxide and adherence to high-carb diet [50] | Глюкокортикостероиды / Glucocorticosteroids [49] |
Текущий стандарт лечения GLUT1DS – кетогенная диета с высоким содержанием жиров, которая повышает уровень кетоновых тел в крови [48]. Кетоны, такие как β-гидроксибутират и ацетоацетат, являются альтернативными, хотя и несовершенными, заменителями глюкозы в обменных процессах нервной системы. Кетоны проникают через гематоэнцефалический барьер (ГЭБ) через переносчик монокарбоксилатов 1 (англ. monocarboxylate transporter 1, MCT1) и служат источником ацетил-коэнзима А (КоА), который в итоге подается в цикл трикарбоновых кислот (ЦТК).
При введении на ранних стадиях заболевания кетогенной диеты уменьшаются клинические проявления эпилептической болезни, однако ее влияние на другие клинические проявления заболевания неоднородно [51]. Этому есть несколько возможных причин. Во-первых, может продолжаться нехватка гликолитических промежуточных продуктов, которые происходят исключительно из глюкозы и потенциально лежат в основе специфических характеристик заболевания. Во-вторых, сама глюкоза и/или GLUT1 служат сигнальными молекулами. Последнее объяснение заключается в быстром снижении уровня белка MCT1 после прекращения грудного вскармливания [52]. Кроме того, длительное лечение кетогенной диетой не только сложно из-за несоблюдения режима, но также может иметь неблагоприятные последствия, включая значительное снижение костной массы, сердечно-сосудистые осложнения из-за атеросклероза и, в редких случаях, индукцию состояния комы.
Инновационным представлялось применение тригептаноина – триглицерида с нечетной цепью (C7), который служит анаплеротическим агентом для пополнения промежуточных продуктов метаболизма ЦТК [53]. Анонсировался препарат как альтернатива кетогенной диете, поскольку он может метаболизироваться как в ацетил-КоА, так и в пропионил-КоА – источник кетоновых тел С3, которые легко проникают через ГЭБ посредством МСТ1 [54]. Однако рандомизированное слепое плацебо-контролируемое клиническое исследование по оценке воздействия препарата на пациентов с GLUT1DS не смогло продемонстрировать его пользу, что позволяет предположить, что тригептаноин имеет ограниченное терапевтическое применение.
Использование более гибких диет, в т.ч. модифицированной диеты Аткинса и ингибиторов карбоангидразы, таких как ацетазоламид или зонисамид, может быть эффективно в более легких случаях [25].
Особое внимание необходимо уделить дополнительному лечению детей, находящихся на кетогенной диете в комплексе с противоэпилептическими препаратами. Комбинации препаратов представлены в таблице 2 [55].
Таблица 2. Современные варианты лечения и меры предосторожности
при синдроме дефицита транспортера глюкозы 1-го типа
(англ. glucose transporter type 1 deficiency syndrom, GLUT1DS)
и эпилепсии (адаптировано по [55])
Table 2. Current treatment options and precautions
for glucose transporter type 1 deficiency syndrom (GLUT1DS)
and epilepsy (adapted from [55])
Терапия / Therapy | Противоэпилептические препараты / Antiepileptic drugs | Препараты, которых следует избегать на фоне диеты / Drug combinations that must be avoided when on a diet | Препараты, которых следует избегать из-за GLUT1DS / Drugs that must be avoided due to GLUT1DS |
Кетогенная диета / Ketogenic diet | Ацетазоламид / Acetazolamide | Вальпроевая кислота / Valproic acid | Фенобарбитал, вальпроевая кислота / Phenobarbital, valproic acid |
Модифицированная диета Аткинса / The modified Atkins diet | Топирамат / Topiramate | Зонисамид / Zonisamide | Ингибиторы тирозинкиназы / Tyrosinekinase inhibitors |
Использование смесей со среднецепочечными триглицеридами / The use of mixtures with medium-chain triglycerides | Зонисамид / Zonisamide | Ацетазоламид / Acetazolamide | Кофеин, этанол / Caffeine, ethanol |
Диета с низким гликемическим индексом / A diet with low glycemic index | Фенитоин / Phenytoin | Топирамат / Topiramate | Диазепам / Diazepam |
Тригептаноин / Triheptanoin | Карбамазепин / Carbamazepine | – | Трициклические антидепрессанты / Tricyclic antidepressants |
Альфа-липоевая кислота / Alpha-lipoic acid | – | – | Общие анестетики, хлоралгидрат / General anesthetics, chloral hydrate |
Поскольку для данной патологии предлагаются диагностические алгоритмы с выработанными критериями, верификация до сих пор остается затруднительной. В связи с этим представляем клиническое наблюдение классического течения заболевания с результатами диагностики и проведенным лечением.
Пациент Р., 19 лет, наблюдался с жалобами на неконтролируемые движения в нижних конечностях в виде «внезапных поднятий обеих ног», в верхних конечностях по типу «вздрагивания рук» в утренние часы до 5 раз в месяц, приступы насильственных движений головой по типу кивков, приступы внезапных падений, приступы потери сознания с судорогами, непроизвольным мочеиспусканием и прикусом языка, преимущественно при побуждении, а также нарушения координации движений.
Ведение пациента осуществлялось сообразно принципам Хельсинкской декларации Всемирной медицинской ассоциации (Форталеза, Бразилия, 2013 г.). От больного получено информированное согласие на проведение клинического обследования и продолженного видео-ЭЭГ-мониторинга. Также получено письменное информированное согласие пациента, разрешающее публикацию истории заболевания и результатов обследования.
Из анамнеза жизни известно, что наследственность по эпилепсии не отягощена. Рожден от первой беременности, протекавшей с угрозой прерывания на ранних сроках. При рождении масса тела 3500 г, рост 52 см.
Самостоятельная ходьба с 2 лет, самостоятельный прием пищи с 2,5 лет, фразовая речь с 3,5 лет, элементарный счет с 6 лет. Матерью отмечается, что в раннем детстве пациент часто ссорился с детьми своего возраста, дрался.
В школу пошел в 7 лет. Учился, со слов матери, «в основном на тройки», со сверстниками общался с недоверием. Окончил 9 классов.
На первом году жизни, по словам родителей, возникли первые приступы в виде адверсий глазных яблок, а в 1,5 года появились спонтанные падения, обусловленные дискоординаций и неустойчивостью при ходьбе. С 2 лет отмечаются вздрагивания, возникающие преимущественно при засыпании и пробуждении, а также кивки, учащающиеся на фоне голода.
В 7 лет выполнена ЭЭГ, обнаружены комплексы «пик – волна» с акцентом в левых лобно-височных отведениях с фотопароксизмальным ответом. После обследования неврологом назначена вальпроевая кислота в дозировке 750 мг/сут – без эффекта. В дальнейшем проводился подбор противоэпилептической терапии – также без значимого эффекта.
Ввиду наличия фармакорезистентности к терапии эпилепсии, задержки нервно-психического развития и выраженных гиперкинезов пациент был направлен на генетическое исследование. В 9 лет генетически верифицирован GLUT1DS: обнаружен гетерозиготный патогенный вариант в гене SLC2A1 (c.177del p.Thr60Argfs*18), что соответствует диагнозу. Поскольку считается, что это аутосомно-доминантная патология, при которой оба родителя здоровы и нет семейного анамнеза, сделан вывод, что имеет место мутация de novo.
С 9 лет введена кетогенная диета, на фоне которой отмечалась значительная положительная динамика состояния пациента, проявлявшаяся в снижении частоты приступов.
С 15 лет возникли хореиформные гиперкинезы в ногах длительностью около 2 ч. В том же году терапия ацетазоламидом и препаратами калия и магния дала положительный эффект в виде уменьшения частоты гиперкинезов.
Зрачки округлой формы, OD=OS. Фотореакции (прямая и содружественная) симметричны. Движения глазных яблок в полном объеме. Язык по средней линии. Надбровный, корнеальный рефлексы живые, симметричные. Глотание не нарушено. При расспросе – умеренная дизартрия. Глоточные и небные рефлексы живые, равные с обеих сторон. Симптомы орального автоматизма отсутствуют. Мышечный тонус в руках и ногах нормальный, симметричный. Во время осмотра пациентом совершаются крупноразмашистые движения верхних конечностей по типу вздрагиваний и плечевого пояса по типу инициации разворота. Мышечная сила 5 баллов, D=S в верхних и нижних конечностях. Глубокие рефлексы: D=S в руках и ногах, живые. Брюшные рефлексы D=S, живые. Нарушений болевой, температурной чувствительности не выявлено. Координаторные пробы выполняет неуверенно, совершает ошибки при выборе конечности, при выполнении – мимопопадание в обеих конечностях. В позе Ромберга неустойчив. Походка неуверенная, с широко расставленными ногами, штампующая. Менингеальных знаков нет. Функции тазовых органов контролирует.
Сознание ясное, ориентирован всесторонне верно. К контакту доступен. Фон настроения неустойчивый, вспыльчив. Эмоционально лабилен. Ажиатирован, невнимателен, игнорирует критические замечания. Выявляется выраженное снижение функции когнитивных процессов в виде нарушения памяти, внимания, отмечается истощаемость. По краткой шкале оценки психического статуса (англ. Mini-Mental State Examination, MMSE) – 21 балл. На момент осмотра без агрессивных и социально опасных тенденций, без продуктивной симптоматики и бредовых идей. Критика к своему состоянию отсутствует.
Проведен анализ концентрации вальпроевой кислоты в крови на фоне приема 500 мг/сут. Результат: 176,25 мкмоль/л (референтный диапазон от 346 до 692 мкмоль/л).
При анализе мочи на фоне кетогенной диеты выявлены: гиперстенурия (1027 г/л), кетонурия (15 ммоль/л), уробилинурия (35 мг/л).
Результаты видео-ЭЭГ-мониторинга дневного сна представлены на рисунке 2.
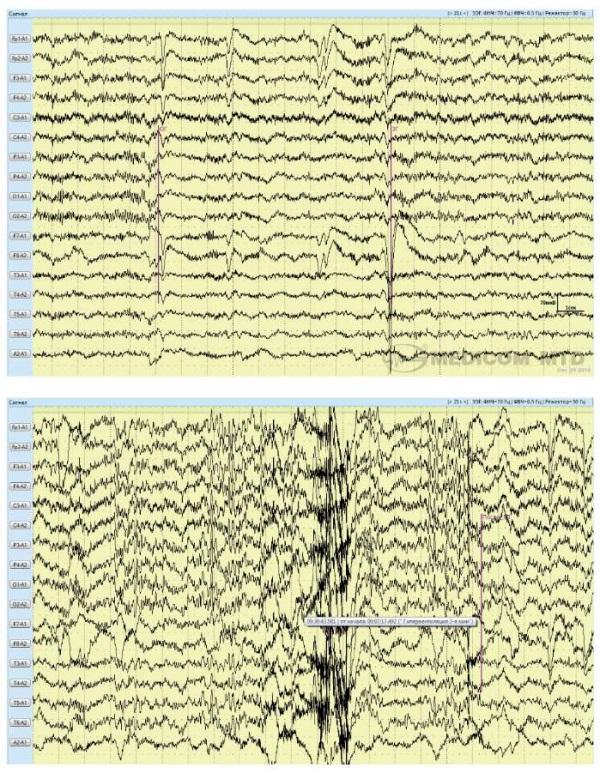
Рисунок 2. Результаты видеоэлектроэнцефалографического мониторинга:
a – на протяжении всей записи регистрируется большое количество неустранимых артефактов,
умеренные диффузные нарушения биоэлектрической активности головного мозга
свидетельствуют о дисфункции стволовых структур с нарушением активации коры,
раздражении коры по всей конвекситальной поверхности, раздражении стволовых структур,
в большей степени на диэнцефальном уровне,
отчетливые локальные нарушения активности не выявляются;
b – на фоне гипервентиляции регистрируются единичные,
умеренные пароксизмальные нарушения активности неспецифического характера
с акцентом в глубоких отделах левого полушария и умеренным вовлечением ствола мозга,
эпилептиформных нарушений активности не отмечено
Figure 2. Video-electroencephalographic monitoring results:
a – a large number of non-recoverable artifacts are recorded throughout the recording;
moderate diffuse disturbances of brain bioelectric activity
evidence about stem structure dysfunction with impaired cortical activation;
cortical irritation throughout the convexital surface; stem structures irritation,
mostly at the diencephalic level; no distinct locally disturbed activity is detected;
b – isolated, moderate paroxysmal activity disorders of non-specific nature
are recorded primarily in the deep parts of the left hemisphere along
with moderately involved brain stem during hyperventilation;
no epileptiform activity disorders are noted
По результатам обследования у пациента выявлены признаки изменения психических процессов по органическому типу. Критика к собственному состоянию снижена (вследствие нарушения функции лобных долей головного мозга). Умеренно выраженные дисфункции кинестетического кистевого и пальцевого гнозиса (неполноценность функционирования постцентральной области левого полушария – черты кинестетической кистевой, пальцевой апраксии), динамического праксиса (неполноценность функционирования премоторной области левого полушария), конструктивного праксиса (неполноценность функционирования теменной доли левого полушария), реципрокной координации (дисфункция теменной доли левого полушария). Умеренно выраженные нарушения оптико-пространственного гнозиса (неполноценность функционирования теменно-затылочной области правого полушария), умеренно выраженные нарушения цветового гнозиса (неполноценность функционирования затылочной доли левого полушария).
Обнаружены умеренно выраженные нарушения экспрессивной и спонтанной речи. Присутствуют элементы дизартрии. Отмечены элементы акалькуляции, умеренно выраженные нарушения абстрагирования и категориального мышления (лобная доля левого полушария). Операциональная сторона мышления нарушена по типу снижения уровня обобщения. Мотивационный компонент мышления нарушен в умеренно выраженной степени по типу разноплановости мышления. Умеренно выраженные нарушения кратковременной, оперативной, долговременной, операциональной памяти (неполноценность функционирования нижней височной доли левого полушария, неполноценность функционирования глубины мозга, в частности гиппокампа). Инфантильность личности, отсутствие мстительности, не замечает оскорблений, отсутствует честолюбие, не обращает никакого внимания на критические замечания. Отсутствуют эгоцентрические побуждения (либо они блокированы). Границы психического образа «Я» не очерчены, отношение к себе не выработано. Формирование мотивации затруднено.
На основании анамнеза заболевания и полученных результатов верифицирован диагноз: «Болезнь де Виво. Генетическая генерализованная эпилепсия. Умственная отсталость».
Ввиду недостаточного эффекта от вальпроевой кислоты и ее несовместимости с кетогенной диетой препарат был постепенно выведен из терапии и назначен клоназепам для купирования гиперкинезов в дозировке 1 мг/сут (0,5 мг утром и 0,5 мг вечером). За период госпитализации приступов и гиперкинезов не отмечалось, общее состояние пациента на момент выписки удовлетворительное. На фоне олигофрении рекомендованы реабилитация, когнитивный тренинг, посещение клинического психолога для оценки динамики нервно-психического развития.
Болезнь де Виво является одной из отличительных форм эпилепсии, которая вызывает сложности в педиатрической практике, в т.ч. и в дифференциальной диагностике между такими нозологиями, как синдром опсоклонус-миоклонус, состояния с судорогами у новорожденных и приобретенная микроцефалия (к примеру, синдром Ангельмана), и другими причинами гипогликорахии, включая состояния, вызывающие хроническую или транзиторную гипогликемию (например, семейный гиперинсулинизм).
Из этого следует, что зачастую ведущим методом диагностики редких неврологических заболеваний является генетический, который позволяет в короткие сроки установить конкретную причину той или иной клинической картины и назначить своевременную адекватную терапию. Представленный случай также демонстрирует, что мутация может возникнуть de novo и семейный анамнез может быть не отягощен, о чем следует помнить неврологу при расспросе больного.
Таким образом, если на прием к неврологу попадает пациент с эпилептическими приступами в анамнезе, которые не купируются назначением антиэпилептической терапии, навязчивыми движениями и задержкой нервно-психического развития, необходимо помнить о таких редких патологиях, приводящих к нарушению углеводного обмена, как болезнь де Виво. Приведенный нами клинический случай показывает, что не все проявления эпилепсии возможно нивелировать назначением противосудорожных препаратов и требуется более тщательный подбор терапии, в т.ч. и немедикаментозной.
1. De Vivo D.C., Trifiletti R.R., Jacobson R.I., et al. Defective glucose transport across the blood-brain barrier as a cause of persistent hypoglycorrhachia, seizures, and developmental delay. N Engl J Med. 1991; 325 (10): 703–9. https://doi.org/10.1056/NEJM199109053251006.
2. Symonds J.D., Zuberi S.M., Stewart K., et al. Incidence and phenotypes of childhood-onset genetic epilepsies: a prospective population-based national cohort. Brain. 2019; 142 (8): 2303–18. https://doi.org/10.1093/brain/awz195.
3. López-Rivera J.A., Pérez-Palma E., Symonds J., et al. A catalogue of new incidence estimates of monogenic neurodevelopmental disorders caused by de novo variants. Brain. 2020; 143 (4): 1099–105. https://doi.org/10.1093/brain/awaa051.
4. Larsen J., Johannesen K.M., Ek J., et al. The role of SLC2A1 mutations in myoclonic astatic epilepsy and absence epilepsy, and the estimated frequency of GLUT1 deficiency syndrome. Epilepsia. 2015; 56 (12): e203–8. https://doi.org/10.1111/epi.13222.
5. Coman D.J., Sinclair K.G., Burke C.J., et al. Seizures, ataxia, developmental delay and the general paediatrician: glucose transporter 1 deficiency syndrome. J Paediatr Child Health. 2006; 42 (5): 263–7. https://doi.org/10.1111/j.1440-1754.2006.00852.x.
6. Seidner G., Alvarez M.G., Yeh J.I., et al. GLUT-1 deficiency syndrome caused by haploinsufficiency of the blood-brain barrier hexose carrier. Nat Genet. 1998; 18 (2): 188–91. https://doi.org/10.1038/ng0298-188.
7. Wang D., Pascual J.M., De Vivo D. Glucose transporter type 1 deficiency syndrome. In: Adam M.P., Feldman J., Mirzaa G.M., et al. (Eds.) GeneReviews®. Seattle (WA): University of Washington, Seattle; 2002.
8. Rotstein M., Engelstad K., Yang H., et al. Glut1 deficiency: inheritance pattern determined by haploinsufficiency. Ann Neurol. 2010; 68 (6): 955–8. https://doi.org/10.1002/ana.22088.
9. Weber Y.G., Storch A., Wuttke T.V., et al. GLUT1 mutations are a cause of paroxysmal exertion-induced dyskinesias and induce hemolytic anemia by a cation leak. J Clin Invest. 2008; 118 (6): 2157–68. https://doi.org/10.1172/JCI34438.
10. Nicita F., Schirinzi T., Stregapede F., et al. SLC2A1 mutations are a rare cause of pediatric-onset hereditary spastic paraplegia. Eur J Paediatr Neurol. 2019; 23 (2): 329–32. https://doi.org/10.1016/j.ejpn.2018.12.004.
11. Yang H., Wang D., Engelstad K., et al. Glut1 deficiency syndrome and erythrocyte glucose uptake assay. Ann Neurol. 2011; 70 (6): 996–1005. https://doi.org/10.1002/ana.22640.
12. Willemsen M.A., Vissers L.E., Verbeek M.M., et al. Upstream SLC2A1 translation initiation causes GLUT1 deficiency syndrome. Eur J Hum Genet. 2017; 25 (6): 771–4. https://doi.org/10.1038/ejhg.2017.45.
13. Pong A.W., Geary B.R., Engelstad K.M., et al. Glucose transporter type I deficiency syndrome: epilepsy phenotypes and outcomes. Epilepsia. 2012; 53 (9): 1503–10. https://doi.org/10.1111/j.1528-1167.2012.03592.x.
14. Wolking S., Becker F., Bast T., et al. Focal epilepsy in glucose transporter type 1 (Glut1) defects: case reports and a review of literature. J Neurol. 2014; 261 (10): 1881–6. https://doi.org/10.1007/s00415-014-7433-5.
15. Peeraer A., Damiano J.A., Bellows S.T., et al. Evaluation of GLUT1 variation in non-acquired focal epilepsy. Epilepsy Res. 2017; 133: 54–7. https://doi.org/10.1016/j.eplepsyres.2017.04.007.
16. Nasser H., Lopez-Hernandez E., Ilea A., et al. Myoclonic jerks are commonly associated with absence seizures in early-onset absence epilepsy. Epileptic Disord. 2017; 19 (2): 137–46. https://doi.org/10.1684/epd.2017.0905.
17. Pearson T.S., Pons R., Engelstad K., et al. Paroxysmal eye-head movements in Glut1 deficiency syndrome. Neurology. 2017; 88 (17): 1666–73. https://doi.org/10.1212/WNL.0000000000003867.
18. Hao J., Kelly D.I., Su J., Pascual J.M. Clinical aspects of glucose transporter type 1 deficiency: information from a Global Registry. JAMA Neurol. 2017; 74 (6): 727–32. https://doi.org/10.1001/jamaneurol.2017.0298.
19. Leen W.G., Taher M., Verbeek M.M., et al. GLUT1 deficiency syndrome into adulthood: a follow-up study. J Neurol. 2014; 261 (3): 589–99. https://doi.org/10.1007/s00415-014-7240-z.
20. Weller C.M., Leen W.G., Neville B.G., et al. A novel SLC2A1 mutation linking hemiplegic migraine with alternating hemiplegia of childhood. Cephalalgia. 2015; 35 (1): 10–5. https://doi.org/10.1177/0333102414532379.
21. Leen W.G., Klepper J., Verbeek M.M., et al. Glucose transporter-1 deficiency syndrome: the expanding clinical and genetic spectrum of a treatable disorder. Brain. 2010; 133 (Pt. 3): 655–70. https://doi.org/10.1093/brain/awp336.
22. Hully M., Vuillaumier-Barrot S., Le Bizec C., et al. From splitting GLUT1 deficiency syndromes to overlapping phenotypes. Eur J Med Genet. 2015; 58 (9): 443–54. https://doi.org/10.1016/j.ejmg.2015.06.007.
23. De Giorgis V., Veggiotti P. GLUT1 deficiency syndrome 2013: current state of the art. Seizure. 2013; 22 (10): 803–11. https://doi.org/10.1016/j.seizure.2013.07.003.
24. Wang D., Pascual J.M., Yang H., et al. Glut-1 deficiency syndrome: clinical, genetic, and therapeutic aspects. Ann Neurol. 2005; 57 (1): 111–8. https://doi.org/10.1002/ana.20331.
25. Tzadok M., Nissenkorn A., Porper K., et al. The many faces of Glut1 deficiency syndrome. J Child Neurol. 2014; 29 (3): 349–59. https://doi.org/10.1177/0883073812471718.
26. Liu Y., Bao X., Wang D., et al. Allelic variations of glut-1 deficiency syndrome: the chinese experience. Pediatr Neurol. 2012; 47 (1): 30–4. https://doi.org/10.1016/j.pediatrneurol.2012.04.010.
27. Atli E.I., Atli E., Yalcintepe S., et al. Customised targeted massively parallel sequencing enables more precise diagnosis of patients with epilepsy. Intern Med J. 2022; 52 (7): 1174–84. https://doi.org/10.1111/imj.15219.
28. Klepper J., Willemsen M., Verrips A., et al. Autosomal dominant transmission of GLUT1 deficiency. Hum Mol Genet. 2001; 10 (1): 63–8. https://doi.org/10.1093/hmg/10.1.63.
29. Klepper J. Absence of SLC2A1 mutations does not exclude Glut1 deficiency syndrome. Neuropediatrics. 2013; 44 (4): 235–6. https://doi.org/10.1055/s-0033-1336015.
30. Hashimoto N., Kagitani-Shimono K., Sakai N., et al. SLC2A1 gene analysis of Japanese patients with glucose transporter 1 deficiency syndrome. J Hum Genet. 2011; 56 (12): 846–51. https://doi.org/10.1038/jhg.2011.115.
31. Liu Y.C., Lee J.W., Bellows S.T., et al. Evaluation of non-coding variation in GLUT1 deficiency. Dev Med Child Neurol. 2016; 58 (12): 1295–302. https://doi.org/10.1111/dmcn.13163.
32. Sánchez-Lijarcio O., Yubero D., Leal F., et al. The clinical and biochemical hallmarks generally associated with GLUT1DS may be caused by defects in genes other than SLC2A1. Clin Genet. 2022; 102 (1): 40–55. https://doi.org/10.1111/cge.14138.
33. Mayorga L., Gamboni B., Mampel A., Roqué M. A frame-shift deletion in the PURA gene associates with a new clinical finding: hypoglycorrhachia. Is GLUT1 a new PURA target? Mol Genet Metab. 2018; 123 (3): 331–6. https://doi.org/10.1016/j.ymgme.2017.12.436.
34. Vaudano A.E., Olivotto S., Ruggieri A., et al. Brain correlates of spike and wave discharges in GLUT1 deficiency syndrome. Neuroimage Clin. 2016; 13: 446–54. https://doi.org/10.1016/j.nicl.2016.12.026.
35. Klepper J., Leiendecker B. GLUT1 deficiency syndrome – 2007 update. Dev Med Child Neurol. 2007; 49 (9): 707–16. https://doi.org/10.1111/j.1469-8749.2007.00707.x.
36. De Vivo D.C., Wang D. Glut1 deficiency: CSF glucose. How low is too low? Rev Neurol. 2008; 164 (11): 877–80. https://doi.org/10.1016/j.neurol.2008.10.001.
37. Leen W.G., Wevers R.A., Kamsteeg E.J., et al. Cerebrospinal fluid analysis in the workup of GLUT1 deficiency syndrome: a systematic review. JAMA Neurol. 2013; 70 (11): 1440–4. https://doi.org/10.1001/jamaneurol.2013.3090.
38. Klepper J. GLUT1 deficiency syndrome in clinical practice. Epilepsy Res. 2012; 100 (3): 272–7. https://doi.org/10.1016/j.eplepsyres.2011.02.007.
39. Leen W.G., de Wit C.J., Wevers R.A., et al. Child neurology: differential diagnosis of a low CSF glucose in children and young adults. Neurology. 2013; 81 (24): e178–81. https://doi.org/10.1212/01.wnl.0000437294.20817.99.
40. Ito Y., Gertsen E., Oguni H., et al. Clinical presentation, EEG studies, and novel mutations in two cases of GLUT1 deficiency syndrome in Japan. Brain Dev. 2005; 27 (4): 311–7. https://doi.org/10.1016/j.braindev.2004.09.010.
41. Hennecke M., Wang D., Korinthenberg R., et al. GLUT1 deficiency syndrome with ataxia, acquired microcephaly and leucoencephalopathy in monozygotic twins. Neuropediatrics. 2005; 36: 140.
42. Veneruzzo G.M., Loos M.A., Armeno M., et al. Glucose transporter type 1 deficiency syndrome: clinical aspects, diagnosis, and treatment. Arch Argent Pediatr. 2023; 121 (1): e202202677. https://doi.org/10.5546/aap.2022-02677.eng.
43. Williams C.A., Angelman H., Clayton-Smith J., et al. Angelman syndrome: consensus for diagnostic criteria. Angelman Syndrome Foundation. Am J Med Genet. 1995; 56 (2): 237–8. https://doi.org/10.1002/ajmg.1320560224.
44. Palladino A.A., Bennett M.J., Stanley C.A. Hyperinsulinism in infancy and childhood: when an insulin level is not always enough. Clin Chem. 2008; 54 (2): 256–63. https://doi.org/10.1373/clinchem.2007.098988.
45. Алифирова В.М., Валикова Т.А., Пугаченко Н.В. и др. Опсоклонус-миоклонус синдром (энцефалопатия Кинсбурна). Бюллетень сибирской медицины. 2019; 18 (4): 233–8. https://doi.org/10.20538/1682-0363-2019-4-233-238.
46. Cantarín-Extremera V., Jiménez-Legido M., Aguilera-Albesa S., et al. Opsoclonus-myoclonus syndrome: clinical characteristics, therapeutic considerations, and prognostic factors in a Spanish paediatric cohort. Neurologia. 2023; 38 (2): 93–105. https://doi.org/10.1016/j.nrleng.2020.04.030.
47. Gataullina S., Lonlay P., Dellatolas G., et al. Topography of brain damage in metabolic hypoglycaemia is determined by age at which hypoglycaemia occurred. Dev Med Child Neurol. 2013; 55 (2): 162–6. https://doi.org/10.1111/dmcn.12045.
48. Klepper J., Leiendecker B. Glut1 deficiency syndrome and novel ketogenic diets. J Child Neurol. 2013; 28 (8): 1045–8. https://doi.org/10.1177/0883073813487600.
49. Guerrini R., De Lorey T.M., Bonanni P., et al. Cortical myoclonus in Angelman syndrome. Ann Neurol. 1996; 40 (1): 39–48. https://doi.org/10.1002/ana.410400109.
50. Aynsley-Green A., Hussain K., Hall J., et al. Practical management of hyperinsulinism in infancy. Arch Dis Child Fetal Neonatal Ed. 2000; 82 (2): F98–107. https://doi.org/10.1136/fn.82.2.f98.
51. Pascual J.M., Ronen G.M. Glucose transporter type i deficiency (G1D) at 25 (1990–2015): presumptions, facts, and the lives of persons with this rare disease. Pediatr Neurol. 2015; 53 (5): 379–93. https://doi.org/10.1016/j.pediatrneurol.2015.08.001.
52. Pellerin L., Pellegri G., Martin J.L., Magistretti P.J. Expression of monocarboxylate transporter mRNAs in mouse brain: support for a distinct role of lactate as an energy substrate for the neonatal vs. adult brain. Proc Natl Acad Sci USA. 1998; 95 (7): 3990–5. https://doi.org/10.1073/pnas.95.7.3990.
53. Mochel F. Triheptanoin for the treatment of brain energy deficit: a 14-year experience. J Neurosci Res. 2017; 95 (11): 2236–43. https://doi.org/10.1002/jnr.24111.
54. Pascual J.M., Liu P., Mao D., et al. Triheptanoin for glucose transporter type I deficiency (G1D): modulation of human ictogenesis, cerebral metabolic rate, and cognitive indices by a food supplement. JAMA Neurol. 2014; 71 (10): 1255–65. https://doi.org/10.1001/jamaneurol.2014.1584.
55. Kass H.R., Winesett S.P., Bessone S.K., et al. Use of dietary therapies amongst patients with GLUT1 deficiency syndrome. Seizure. 2016; 35: 83–7. https://doi.org/10.1016/j.seizure.2016.01.011.
Шова Наталья Игоревна – к.м.н., старший научный сотрудник отделения лечения больных с экзогенно-органическими расстройствами и эпилепсией.
ул. Бехтерева, д. 3, Санкт-Петербург 192019
WоS ResearcherID AAI-3755-2020; Scopus Author ID 57215893698
Михайлов Владимир Алексеевич – д.м.н., главный научный сотрудник и научный руководитель отделения лечения больных с экзогенно-органическими расстройствами и эпилепсией и отделения реабилитации пациентов с психосоматическими расстройствами.
ул. Бехтерева, д. 3, Санкт-Петербург 192019
WоS ResearcherID B-3272-2017
Романюго Григорий Дмитриевич – врач-ординатор отделения лечения больных с экзогенно-органическими расстройствами и эпилепсией.
ул. Бехтерева, д. 3, Санкт-Петербург 192019
Шова Н.И., Михайлов В.А., Романюго Г.Д. Болезнь де Виво (сочетание миоклонико-астатической эпилепсии и хореи): литературный обзор, описание клинического наблюдения. Эпилепсия и пароксизмальные состояния. 2024;16(2):145-156. https://doi.org/10.17749/2077-8333/epi.par.con.2024.182
Shova N.I., Mikhailov V.A., Romanyugo G.D. De Vivo disease (myoclonic-astatic epilepsy combined with chorea): literature review, clinical case description. Epilepsy and paroxysmal conditions. 2024;16(2):145-156. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2024.182

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































