


ORIGINAL ARTICLES
Background. In case of ineffective conservative antiepileptic therapy, surgical treatment aimed at removing the epileptogenic focus may be applied. Resection procedures allow to eliminate seizures in most patients, but in 20–30% cases they persist or recur, thereby proposing to use some neuromodulation.
Objective: to assess effectiveness of neuromodulation in patients with drug-resistant epilepsy (DRE) after failed resection surgical interventions.
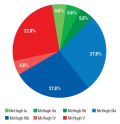
Material and methods. A retrospective data analysis was carried out involving 23 DRE patients who had undergone vagus nerve stimulation (VNS) or deep brain stimulation (DBS) of the anterior nucleus of the thalamus (ANT) or hippocampus (HP) after failed surgeries. The VNS system was implanted in 18 (78.3%) patients, the HP-DBS system – in 3 (13.0%), and the ANT-DBS system – in 2 (8.7%). The results after surgical interventions were assessed according to the Engel scale, VNS therapy – by the McHugh (MH) scale, DBS therapy – by the degree of reduced seizure rate as a percentage. The average follow-up was 56.5 months.
Results. Patients with implanted VNS system were found to have the outcome presented as MH Ia–IIb in 3 (16.7%) cases, MH IIIa–IIIb in 10 (55.5%) cases, MH IV–V in 5 (27.8%) cases. In HP-DBS group, 2 out of 3 patients showed a decline in seizure rate by more than 50% from the baseline level, and 1 patient experienced an improvement in seizure severity. In the ANT-DBS group, one patient had a 60% reduction in seizure rate and an improvement in seizure severity, another one showed no change in seizure rate.
Conclusion. Neuromodulation in DRE patients can significantly lower seizure rate in more than half of patients after failed surgical treatment.

Objective: to investigate the effects of sodium valproate on plasma concentrations of homocysteine, folate and vitamin B12 levels in epileptic patients with long-standing tonic-clonic seizures compared to newly diagnosed epileptic patients and healthy controls.
Material and methods. The study included 90 participants (mean age 36.30±12.83 years, the majority (58.89%) were males) divided into three groups: 30 non-epileptic people (control Group 1), 30 newly diagnosed epileptic patients (Group 2), and 30 patients with long-term tonic-clonic seizures epilepsy (Group 3). In Group 3, patients received sodium valproate therapy. All subjects underwent clinical and neurological examinations. Differences in plasma levels of homocysteine, folic acid and vitamin B12 in three groups were investigated after 6 months of follow-up.
Results. Homocysteine level in Groups 2 and 3 was increased; for Group 2 it was significantly higher than for Groups 3 and 1 (p=0.001). Plasma folate level in Groups 2 and 3 was decreased; for Group 3 it was significantly higher than for Group 2 and lower than for Group 1 (p=0.001). Vitamin B12 level in Groups 2 and 3 was decreased, but the difference was not significant (p=0.090). In Groups 1 and 2, a significant correlation was observed between the indicators.
Conclusion. Sodium valproate аdministration might disrupt the homeostatic level of homocysteine, folate and vitamin B12 and cause irregularity of their plasma contents in epileptic patients with long-standing tonic-clonic seizures.
Background. Lithium salts are used in psychiatry and may exhibit neuroprotective effects due to anion constituents.
Objective: to study a dose-dependent effect of oral lithium ascorbate (LiAsc) and lithium carbonate on manifestation and severity of primary generalized seizures caused by thiosemicarbazide in rats in vivo.
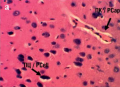
Material and methods. The study was conducted on 30 Wistar white male rats weighing 200–300 g in five groups: control, lithium carbonate 5 mg/kg, LiAsc 5, 10, 15 mg/kg/day, administered by probing for 14 days. The model of primary generalized seizures was reproduced by intraperitoneally administered 28 mg/kg thiosemicarbazide. The degree of neurological deficit was assessed based on seizure indicators (latency period before seizures, number of seizures and startles, clonic seizures, etc.). Histopathological studies of brain tissue were carried out using morphometry of histological sections on BioVision image analyzer (Austria).
Results. A course of carbonate and LiAsc significantly reduced seizure duration. The use of LiAsc at doses of 10–15 mg/kg/day not only significantly shortened seizure duration (p=0.01), but also increased the latent period before seizures (p=0.03), reduced the number of seizures (р<0.05) as well as rate of developing tonic extension (р=0.01).
Conclusion. LiAsc at dose of 10 mg/kg/day is sufficient for reducing neurological deficits in thiosemicarbazide-induced seizure model, further confirmed by the data of brain histopathological examination and morphometric analysis.
CLINICAL CASES
PACS1 neurodevelopmental disorder (Schuurs-Hoeijmakers syndrome; MIM #615009) is a rare autosomal dominant genetic syndrome characterized by developmental delay, intellectual disability, dysmorphic features, and rare seizures. The article describes a clinical case of PACS1 syndrome in a female patient with developmental delay, speech disorder, motor development delay and epilepsy coupled to described variants in PACS1 gene (rs398123009, chr11:6621120, c.607C>T, p.Arg203Trp). Knowing PACS1 syndrome molecular mechanisms is important not only for genotype-phenotypic correlation, but also for developing new therapeutic approaches that could improve the quality of patients’ life.
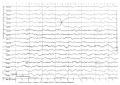
A clinical observation of a patient with Dravet syndrome caused by SCN1A gene mutation is presented. Dravet syndrome is a severe epileptic encephalopathy that occurs in early childhood, accompanied by seizure polymorphism, drug-resistant course and severe cognitive impairment. The current clinical case confirms the opportunity for drug controlled course of Dravet syndrome. A two-year remission was achieved using polytherapy with antiepileptic drugs. Currently, a complete remission during duotherapy with topiramate in combination with levetiracetam is sustained. The described clinical case also demonstrates preservation of cognitive functions: the child successfully acquires full general education program. It should be noted that upon early relief of epileptic seizures, no cognitive functions become affected.
Transient global amnesia (TGA) and transient epileptic amnesia (TEA) are rare phenomena in clinical practice that manifest as transient cognitive amnestic impairments. Despite the similarity in clinical picture, such conditions are pathogenetically heterogeneous and require different therapeutic approaches. TGA is a clinical syndrome characterized by sudden anterograde amnesia of the event lasting up to 24 hours, lacking focal neurological symptoms, and not prone to recurrence. Mimicking TGA, TEA often occurs manifested as epileptic seizures with impaired awareness of varying duration, including long-term (more than 24 hours), as a variant of focal epilepsy. TEA is characterized by recurrent episodes, combination with other manifestations of epilepsy, and comorbidity with neurodegenerative diseases (dementia). For differential diagnosis, it is necessary to use prolonged video-electroencephalographic monitoring with sleep recording, neuroimaging methods (brain magnetic resonance imaging, positron emission tomography), psychological testing, biochemical examination for markers of neurodegeneration.
SCIENTIFIC SURVEYS

De Vivo disease is a rare genetic disorder associated with glucose transporter type 1 (GLUT1) deficiency. We provide a review of publications describing various clinical manifestations of this syndrome, including the combination of epilepsy with choreic hyperkinesis. The seizures related to De Vivo disease are resistant to basic antiepileptic therapy. The ketogenic diet is suggested as the main treatment method. We present our own clinical observation describing an 18-years old male patient, who had myoclonic-astatic seizures and atactic manifestations such as impaired movements coordination and walking instability, which first appeared at the age of 1.5 years. Due to therapy with valproic acid drugs, seizures persisted with a frequency of up to 5 times a month. From the age of 17, involuntary violent irregular movements of trunk and limb muscles emerged, occurring at a fast pace. The patient underwent a comprehensive examination; as a result, the diagnosis of De Vivo disease was verified. By prescribing proper antiepileptic therapy and ketogenic diet, it was possible to stabilize the patient's condition and stop De Vivo disease-related manifestations of epilepsy and hyperkinetic disorder. We pay attention to the differential diagnosis of conditions characterized by epileptic seizures, mental retardation and violent movements, as well as to the diagnosis and management tactics of patients with De Vivo disease. Unfortunately, not all patients with this pathology receive adequate pathogenetic and symptomatic therapy often undergoing numerous hospitalizations, since the major cause underlying such symptoms, namely GLUT1 deficiency, is not diagnosed.

Epilepsy belongs to one of the most ancient diseases. From the days of yore, patients suffering from epilepsy faced stigma and discrimination because medical knowledge at that time was limited so that such condition was associated with various mystical and mythical phenomena, with no treatment available. The article extensively examines the issue of epilepsy stigma, covering the long history of the disease and highlighting stigma as a serious social problem. Many epileptic patients suffer not only from the disease symptoms but also from social discrimination, which profoundly lowers their quality of life and leads to social maladjustment. Unfortunately, epilepsy stigma has not disappeared completely and persists not only in developing but also in developed countries. We demonstrate the relevance of the problem and the need to increase relevant awareness. The times when epileptic patients were considered possessed by evil spirits and incurable are long gone, so any form of social discrimination against such people is unacceptable in modern society.

This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.
ISSN 2311-4088 (Online)



































