



Contents
Scroll to:
https://doi.org/10.17749/2077-8333/epi.par.con.2024.180
Scroll to:
A clinical observation of a patient with Dravet syndrome caused by SCN1A gene mutation is presented. Dravet syndrome is a severe epileptic encephalopathy that occurs in early childhood, accompanied by seizure polymorphism, drug-resistant course and severe cognitive impairment. The current clinical case confirms the opportunity for drug controlled course of Dravet syndrome. A two-year remission was achieved using polytherapy with antiepileptic drugs. Currently, a complete remission during duotherapy with topiramate in combination with levetiracetam is sustained. The described clinical case also demonstrates preservation of cognitive functions: the child successfully acquires full general education program. It should be noted that upon early relief of epileptic seizures, no cognitive functions become affected.
Abusueva B.A., Shanavazova M.D., Askevova М.A., Khalilov V.S., Bobylova M.Yu. Atypical course of severe myoclonic epilepsy of infancy (Dravet syndrome). Epilepsy and paroxysmal conditions. 2024;16(2):130-136. https://doi.org/10.17749/2077-8333/epi.par.con.2024.180
Convulsive syndrome holds an important place among pathological conditions in young children comprising 10% all emergency requests for medical help [1]. There are several major causes resulting in convulsive syndrome including infectious and inflammatory diseases of the central nervous system, brain malformations, hyperthermic syndrome, metabolic disorders, etc. [2][3].
Febrile seizures in pediatric population have the incidence of 2–4%. Simple (typical) febrile seizures lasting less than 15 minutes are not accompanied by focal neurological symptoms, whereas duration of complex febrile seizures exceeds 15 minutes being clinically characterized by focal symptoms [3][4].
Dravet syndrome (DS) is a severe myoclonic epilepsy in infancy, which is first manifested by febrile and afebrile generalized and focal epileptic seizures. In typical cases, myoclonic seizures are mandatory for DS clinical picture characterized by mental retardation and resistance to antiepileptic drugs (AEDs) [5–7]. According to the 2017 International League Against Epilepsy (ILAE) classification, DS belongs to the genetic forms of epilepsy with febrile seizures plus [5]. The majority (80%) of DS patients carry a mutation in the SCN1A gene [8]. Various studies report that DS incidence rate comprises one case per 20–40 thousand population, at 2:1 boys-to-girls ratio [7].
DS most often begins in the first year of life peaking at age of month 5–6 old. Sometimes, its onset may coincide with acute respiratory viral infection (ARVI) or during post-vaccination period, but it is essential to understand that it does not necessarily suggest a direct link between vaccination and pathology. Emergence of post-vaccination disease may be associated with other factors, which has been extensively investigated to gain deeper insights into all relevant aspects.
The course of the disease can be divided into three stages: i) till the age of 1 year old, the stage of debut, the period of clonic mainly febrile seizures; ii) catastrophic stage at the age of 1–4 years old, with febrile and afebrile seizures observed (i.e., occurring during child’s somatic well-being) such as focal motor seizures, myoclonic, atypical absences, alternating hemiclonias, and tonic-clonic seizures. However, epileptic myoclonus (active and negative) is the key sign in the clinical picture. All seizures tend to have a status course (30 minutes or more) and are resistant to AED therapy [6]. From the moment the seizure frequency elevates, a marked delay in child’s mental development with further regression of previously acquired psycho-speech skills is noted, whereas motor development remains not much impaired [11]; iii) 4–5 years after the onset, it is characterized as a stabilization stage, with seizure frequency decline and dominance of severe cognitive and neurological impairments [5][6].
An electroencephalographic (EEG) study reveals slowed basal activity, as well as a dominance of 4–5 Hz theta rhythm. In interictal period, diffuse and regional epileptiform activity is recorded in 70% cases. Ictal EEG in 50% patients is characterized by photosensitivity, and bright light may occasionally result in self-induced seizures. Epileptiform activity can be triggered by eye closure, photosensitivity, and tends to aggravate during sleep. It should be noted that at least 1–2-year timeframe may lie between disease onset and its diagnosis [6].
The main criteria for DS diagnostics are as follows [12]:
A differential diagnosis should be performed between DS and developmental and epileptic encephalopathy (DEE) caused by PCDH19 gene mutation, which is accompanied by epileptic seizures and intellectual disability, but solely limited to the female gender. Clinical picture in some patients with such diseases is characterized by febrile and afebrile seizures in infancy, a serial and status paroxysmal course during fever as well as post-onset developmental regression [13]. Because both diseases show similar clinical presentations, testing for mutations in the PCDH19 gene is required for patients with clinical features of DS and negative mutant SCN1A gene data [9].
Epilepsy in DS is characterized by a drug-resistant course. It is important to avoid body temperature rise [14]. Valproates are considered as the drugs of choice for DS treatment [15] known to be primarily effective in atypical absence seizures and epileptic myoclonus. Valproate is used at a dose of 30–80 mg/kg/day. Topiramate is particularly effective in DS especially in case of generalized convulsive seizures and alternating hemiconvulsions. The drug is applied starting at dose of 12.5 mg/day with a gradual 12.5 mg per week increase up to a dose of 50–200 mg/day in 2 divided doses [15]. Sodium channel blockers (carbamazepine, lamotrigine, phenytoin, vigabatrin) are contraindicated in DS because have a high risk of primarily myoclonic seizure aggravation [16][17]. If a monotherapy is ineffective, AEDs are used in combinations, of which the most preferable is topiramate + valproate. Another effective combination is presented by topiramate + levetiracetam or valproate + succinimides. Ethosuximide is administered at a dosage of 250–750 mg/day in 2–3 doses [15].
M.Yu. Bobylova [6] reported the experience of using perampanel for DS in two representative cases underlining its high effectiveness as an adjunctive therapy: female patient 1 had achieved a three-year-long drug remission regarding all seizures, whereas patient 2 had afebrile attacks blocked, but during ARVI only febrile attacks were observed. Other treatment options (corticosteroids, immunoglobulins, ketogenic diet, vagus nerve stimulation) revealed low effectiveness in DS [6].
To combat photosensitivity and self-induced paroxysms, it is recommended to wear glasses with blue lenses. It is crucial to prevent body temperature rise, and in case of hyperthermia, urgent use of antipyretics is required. Hot baths and overheating should be avoided [6][18].
DS has an unfavorable prognosis most often coupled to profound disability. Remission, usually short-term, occurs in rare cases [15][19]. Consequently, the major treatment goals are to lower seizure rate, prevent the serial and status course, and improve cognitive functions. Apart from epilepsy, DS is a developmental and epileptic encephalopathy, i.e. its clinical picture also includes intellectual disorders largely resulting in social maladjustment [11]. In adolescence, seizures become less frequent, but the neurological status is dominated by extrapyramidal disorders and severe cognitive impairment.
DS mortality in infancy is about 20% [6][7]. In adult patients, one of the common causes of death in sleep is sudden unexpected death in epilepsy (SUDEP) followed by status epilepticus [20].
Here, there is presented a clinical case of a female child (patient S., born in 2015) with severe epilepsy in infancy (Dravet syndrome).
The child is from the first pregnancy, delivery proceeded without pathology. After birth, patient S. screamed immediately, with an Apgar score of 8–9 points. Until the first year of age, patient S. developed according to age. From birth, an impaired thermoregulation was noted; the body temperature increased when the child was in warm clothes in hot weather. Patient S. poorly tolerated the sun, but relatively well felt in hot bath and shower. No familial history of epilepsy was recorded.
The first seizure occurred at 6 months during sleep – lasting 10–15 minutes with myoclonic twitching during hyperthermia. Then the seizures recurred at age of 10 and 12 months, also along with fever, but with a status course of 40–45 minutes and development of postictal Todd's paralysis stopped with Relanium. Later, both febrile and afebrile seizures emerged.
After the age of 1 year old, neurological deficits were noted in the form of hypotonia, unsteady gait, and delayed psycho-speech development, with EEG-recorded epileptiform activity: single sharp-slow wave complexes were observed in the right temporo-occipital and left fronto-central regions.
Whole exome sequencing was performed on the NextSeq 500 platform (lllumina, USA) in the Genomed laboratory (Russia). DNA sequencing (Hereditary Epilepsies panel) data detected a heterozygous mutation in exon 6 of the SCN1A gene resulting in amino acid deletion at position 318.
At the age of 10 months, it was attempted to treat patient S. with valproic acid drops for oral administration, then in microgranules – lacking any effect, with seizure recurrence. A positive effect was observed after administering topiramate at a dose of 25 mg twice a day in combination with valproate at a dose of 100 mg twice a day.
At the age of 18 months, a personalized rehabilitation program was developed due to complaints of unsteady gait, frequent falls while walking, decreased muscle tone, speech delay, decreased concentration, lack of developed play activities, and emotional lability. The program included classes with a speech therapist, physical therapy, physiotherapy (thermal procedures, electrophoresis, paraffin therapy for distal extremities), and swimming pool lessons.
At the age of 2 years, seizures emerged as freezing, and ultrasound (US) examination revealed hepato- and splenomegaly, paralleled with altered biochemical blood parameters defined as verse events. Therefore, patient S. was switched from using valproate to levetiracetam at a dose of 180 mg twice a day, whereas topiramate dosage was increased up to 50 mg twice a day. Dose escalation was coupled to decline in seizure rate lasting several weeks.
At the age of 2 years 4 months, condition of patient S. deteriorated by showing three status seizures within a month. Two 40–45-minute-long seizures and a single febrile attack, which could not be stopped for more than 2 hours, were recorded. For this reason, patient S. was transported by an ambulance to the diagnostic department of Kuraev Children's Republican Clinical Hospital (Republic of Dagestan).
At admission, patient S. was determined to be in serious condition. At seizure end, the child had nausea and vomiting, positive meningeal symptoms (a positive neck stiffness symptom (+3cm), Kernig's sign 140–150 degrees). In this regard, patient S. was punctured, but no significant changes were found in the cerebrospinal fluid. After lumbar puncture, condition of patient S. improved, and symptoms regressed. A dose of topiramate was decided to increase up to 75 mg twice a day.
Over the next 3 years of life, seizures resulted again from fever during ARVI and about two or three of them occurred in physical health. The entire follow-up period was monitored by ultrasound as well as liver and kidney biochemical parameters: after valproate was replaced with levetiracetam, all laboratory data were within normal ranges.
At the age of 6 years, patient S. had seizure remission while taking AEDs, continuing to attend classes with a speech therapist/pathologist on neurosensory development, classes on neurodynamic gymnastics, as well as swimming pool lessons. Massage courses excepting the cervical-collar area and head were applied, and micropolarization as well as thermal procedures are performed.
Upon seizure remission, a marked progress in psychomotor and speech development was noted while using physical rehabilitation methods, attending classes with a psychologist, speech therapist, sensory integration, and neurogymnastics. Despite the severely disabling diagnosis, by the age of 5 patient S. acquired many new skills and knowledge, uses interactive toys, and assembles complex structures. By the age of 8 years, patient S. went to school, successfully and fully mastering the general education program, having neatness and self-care skills fully developed, and helps in caring for younger sibling brother.
2018 video-EEG monitoring data recording regional epileptiform activity on the left occipitotemporal lead are presented in Figure 1. In 2023, the main rhythm was shown to be within age range, without epileptiform activity.
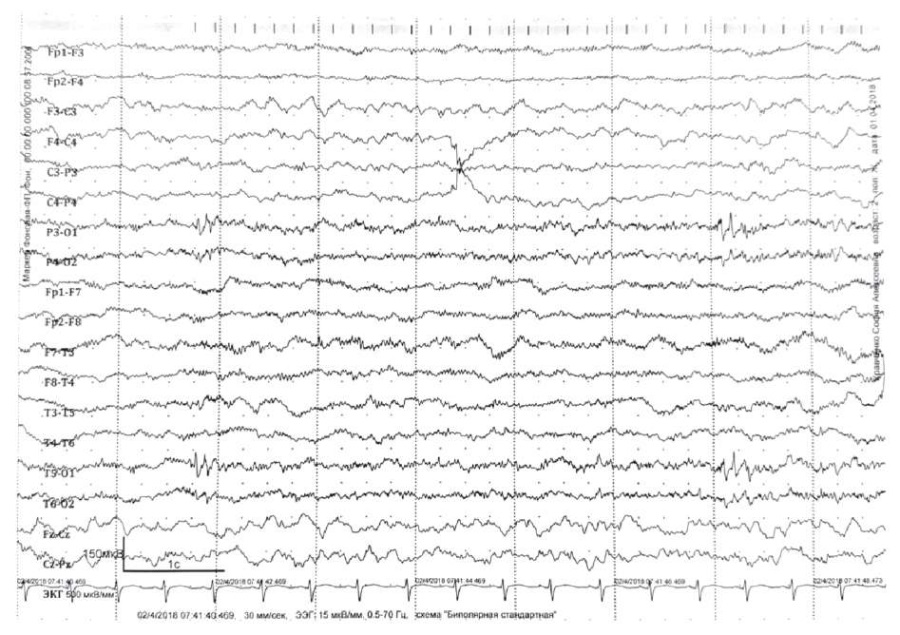
Figure 1. The patient’s electroencephalogram dated 2018.
Regional epileptiform activity – polyspike discharge
in the left posterior-temporal and parietal-central zones, amplitude up to 120 μV
Рисунок 1. Электроэнцефалограмма пациентки от 2018 г.
Региональная эпилептиформная активность – разряд полиспайков
в левой задне-височной и теменно-центральной зонах амплитудой до 120 мкВ
Dravet syndrome is a hard-to-control epileptic syndrome [14], which debuts in the first year of life in a child with normal medical history and psychomotor development. At the onset, seizures usually febrile-provoked may be short or long-lasting [5][7][9]. In the current case, the patient’s seizures were first recorded at the age of 6 months during hyperthermia.
Febrile seizures occur even at low body temperature, of focal pattern; mainly being represented as alternating hemiclonic seizures with transformation into status epilepticus [10]. Here, a status course was also observed in patient S. The persistent seizures were characterized by polymorphism – bilateral tonic-clonic seizures, serial myoclonus, focal seizures with subsequent secondary generalization and resistance to AED therapy.
E.D. Belousova et al. reported that DS patients develop pyramidal insufficiency in the form of hyperreflexia, diffuse muscle hypotonia along with motor clumsiness, and ataxia [9]. Above 7 years of age, extrapyramidal disorders emerged (hypokinesia, tremor, increased plastic muscle tone) [5], with orthopedic complications arising at puberty (kyphoscoliosis, flat feet) [6].
In the current case, patient S. developed normally by the age of up to one year old, not contradicting available publications. Above one year of age, patient S. had a neurological deficit showed as hypotonia, unsteady gait, and delayed psycho-speech development. After focal neurological symptoms emerged at the age of 18 months, patient S. received the necessary amount of rehabilitation treatment, taking into account relevant indications and contraindications. In addition, it was possible to stop epileptic seizures. In our opinion, it allowed to compensate for developmental delay.
Regional epileptiform activity of patient S. was EEG recorded at one year of age, which correlated with deteriorated clinical picture. Video-EEG monitoring of patient S. at the age of 8 years demonstrates the age-related normal range for basic rhythm and the lack of epileptiform activity. Previously, it was reported that DS patients have EEG-based slowed basic activity dominated by 4–5 Hz theta rhythm. Diffuse and regional epileptiform activity is recorded in 70% cases during interictal period increasing during sleep, with characteristic photosensitivity [6].
It was noted above, topiramate is effective in generalized convulsive seizures and alternating hemiconvulsions during DS, being prescribed starting from 12.5 mg/day by gradually escalating dose of 12.5 mg per week up to 50–200 mg/day administered in two doses [15].
In the current clinical case, a positive result was observed precisely after using topiramate. Patient S. also received valproate, which resulted in alleviated seizures, but adverse events developed. Valproate was gradually withdrawn and switched to levetiracetam. At the age of 6 years, clinical seizure remission was achieved by combining AEDs (levetiracetam and topiramate). While writing the manuscript, complete clinical and electroencephalographic remission in patient S. was recorded, and the former has persisted for 30 months.
The presented clinical case demonstrates a feasibility of prescribing AED polytherapy in DS patients aimed at rapidly achieving and maintaining remission and, as a consequence, improving the disease outcome, as well as ameliorating the severity of cognitive impairment. The rehabilitation methods used here demonstrated safety and effectiveness.
Patient S. suffering from DS and described here proves that a favorable outcome may be achieved in the case of successful and rapid blockade of epileptic seizures, as well as during rehabilitation and habilitation measures, with compliance of a patient, his/her legal representative and a doctor. This casts doubt on the certainty of an unfavorable outcome in Dravet syndrome. Our observation is unique in its kind; rare cases of Dravet syndrome with similar course are documented in the world publications.
1. Karlov V.A. Step-by-step therapy of epileptic status. General recommendations on doses and administration regimens. Мoscow; 2009: 3 (in Russ.).
2. Dolinina A.F. Clinical and genetic characteristics of the Dravet syndrome. Pediatric Bulletin of the South Ural. 2017; 2: 37–40 (in Russ.).
3. Kholin A.A., Ilyina E.S., Kolpakchi L.M., et al. Malignant migrating partial seizires of infancy. Clinical observation of 6 cases. Russian Journal of Child Neurology. 2007; 2 (2): 25–38 (in Russ.).
4. Hernandez M., Pedraza M., Mesa T., Troncoso M. Complex febrile seizures or Dravet syndrome? Description of 3 case reports. ReV Chil Pediatr. 2014; 85 (5): 588–93 (in Spanish). https://doi.org/10.4067S0370-41062014000500010.
5. Bobylova M.Yu. Resistant epilepsy forms, associated with fever-provoked (febrile) epileptic seizures (Dravet syndrome, DESC, HHE) (a lecture). Russian Journal of Child Neurology. 2012; 7 (4): 31–40 (in Russ.).
6. Bobylova M.Yu. The experience of using perampanel in Dravet syndrome on the example of two cases. Vestnik epileptologii / Bulletin of Epileptology. 2020; 1: 20–5 (in Russ.).
7. Khuzina G.R., Zakirova D.R., Tukhbatullina E.L., Iksanova E.N. Dravet syndrome. Vestnik soVremennoi klinicheskoi mediciny / The Bulletin of Contemporary Clinical Medicine. 2013; 6 (Suppl. 1): 49–52 (in Russ.).
8. Rosander C., Halböök T. Dravet syndrome in Sweden: a population-based study. DeV Med Child Neurol. 2015; 57 (7): 628–33. https://doi.org/10.1111/dmcn.12709.
9. Belousova E.D., Sharkov A.A. Difficulties in the diagnosis, prognosis and treatment of genetic epileptic encephalopathies: the view of a neurologist. Zhurnal neVrologii i psikhiatrii imeni S.S. KorsakoVa. 2019; 119 (11-2): 34–40 (in Russ.). https://doi.org/10.17116/jnevro201911911234.
10. Mukhin K.Yu., Petrukhin A.S., Kholin A.A. Epileptic encephalopathies and similar syndromes in children. Мoscow: Art-Servis Ltd.; 2011: 157–74 (in Russ.).
11. Dombrovskaya E.A., Pugolovkin K.A. Dravet syndrome – severe myoclonic epilepsy of infancy: experience of 2-year remission in a child aged 2 years 7 months. Pediatria. 2016; 95 (5): 168–71 (in Russ.).
12. Guerrini R. Dravet syndrome: the main isues. Eur J Paediatr Neurol. 2012; 16 (Suppl. 1): S1–4. https://doi.org/10.1016/j.ejpn.2012.04.006.
13. Mukhin K.Yu., Pylaeva O.A., Dolinina A.F., et al. Epilepsy caused by PCDH19 gene mutation: a review of literature and the authors’ observations. Russian Journal of Child Neurology. 2016; 11 (2): 26–32 (in Russ.). https://doi.org/10.17650/2073-8803-2016-11-2-26-32.
14. Abril Jaramillo J., Estevez Maria J.C., Girón Úbeda J.M., et al. Effectiveness and safety of perampanel as early add-on treatment in patients with epilepsy and focal seizures in the routine clinical practice: Spain prospective study (PERADON). Epilepsy BehaV. 2020; 102: 106655. https://doi.org/10.1016/j.yebeh.2019.106655.
15. Mukhin K.Yu., Bobylova M.Yu., Chadaev V.A., Petrukhin A.S. Epileptic syndromes. Diagnosis and therapy. 4th ed. Мoscow: Binom; 2018: 608 pp. (in Russ.).
16. Wallace A., Wirrell E., Kenney-Jung D.L. Pharmacotherapy for Dravet syndrome. Paediatr Drugs. 2016; 18 (3): 197–208. https://doi.org/10.1007/s40272-016-0171-7.
17. Wu Y.W., Sullivan J., McDaniel S.S., et al. Incidence of Dravet syndrome in a US population. Pediatrics. 2015; 136 (5): e1310–5. https://doi.org/10.1542/peds.2015-1807.
18. Brunklaus A., Zuberi S.M. Dravet syndrome – from epileptic encephalopathy to channelopathy. Epilepsia. 2014; 55 (7): 979–84. https://doi.org/10.1111/epi.12652.
19. Kang J.Q., Macdonald R.L. Molecular pathogenic basis for GABRG2 mutations associated with a spectrum of epilepsy syndromes, from generalized absence epilepsy to Dravet syndrome. JAMA Neurol. 2016; 73 (8): 1009–16. https://doi.org/10.1001/jamaneurol.2016.0449.
20. Cooper M.S., Mcintosh A., Crompton D.E., et al. Mortality in Dravet syndrome. Epilepsy Res. 2016; 128: 43–7. https://doi.org/10.1016/j.eplepsyres.2016.10.006.
Burliat A. Abusueva – MD, PhD, Associate Professor, Chief of Chair of Nervous Diseases, Medical Genetics and Neurosurgery, Dagestan State Medical University.
1 Lenin Sq., Makhachkala 367000, Republic of Dagestan
Mukminat D. Shanavazova – Assistant Professor, Chief of Chair of Nervous Diseases, Medical Genetics and Neurosurgery, Dagestan State Medical University.
1 Lenin Sq., Makhachkala 367000, Republic of Dagestan
Mariam A. Askevova – Assistant Professor, Chief of Chair of Nervous Diseases, Medical Genetics and Neurosurgery, Dagestan State Medical University.
1 Lenin Sq., Makhachkala 367000, Republic of Dagestan
Varis S. Khalilov – MD, PhD, Associate Professor, Badalyan Chair of Neurology, Neurosurgery and Medical Genetics, Faculty of Pediatrics, Pirogov Russian National Research Medical University; Radiologist, Department of Magnetic Resonance Imaging, Federal Scientific Center for Children and Adolescents, FMBA of Russia.
1 Ostrovityanov, Moscow 117997; 20 Moskvorechye Str., Moscow 115409
Maria Yu. Bobylova – MD, PhD, Associate Professor, Neurologist-Epileptologist, Svt. Luka's Institute of Child Neurology and Epilepsy.
8 Nagornaya Str., Moscow 108842
Abusueva B.A., Shanavazova M.D., Askevova М.A., Khalilov V.S., Bobylova M.Yu. Atypical course of severe myoclonic epilepsy of infancy (Dravet syndrome). Epilepsy and paroxysmal conditions. 2024;16(2):130-136. https://doi.org/10.17749/2077-8333/epi.par.con.2024.180

117042, Moscow, Chechersky passage, 24
Tel.: +7(495)6495495
e-mail: info@irbis-1.ru



































