



Содержание
Перейти к:
https://doi.org/10.17749/2077-8333/epi.par.con.2024.183
Перейти к:
Синдром нарушения развития нервной системы PACS1 (синдром Схюрс-Хоймакерса (англ. Schuurs-Hoeijmakers syndrome); MIM 615009) – редкое аутосомно-доминантное генетическое заболевание, характеризующееся задержкой развития, интеллектуальным дефицитом, дисморфическими чертами, а иногда и судорогами. В статье представлен клинический случай синдрома PACS1 у пациентки с задержкой психоречевого и моторного развития, эпилепсией и описанными вариантами в гене PACS1 (rs398123009, chr11:6621120, c.607C>T, p.Arg203Trp). Знание молекулярных механизмов развития синдрома PACS1 важно не только для генотип-фенотипической корреляции, но и для разработки новых терапевтических подходов в лечении, которые могли бы улучшить качество жизни пациентов.
Кожанова Т.В., Жилина С.С., Мещерякова Т.И., Михайлова А.Д., Крапивкин А.И., Заваденко Н.Н. Редкий генетический синдром Схюрс-Хоймакерса (синдром PACS1). Эпилепсия и пароксизмальные состояния. 2024;16(2):120-129. https://doi.org/10.17749/2077-8333/epi.par.con.2024.183
Kozhanova T.V., Zhilina S.S., Meshcheryakova T.I., Mikhailova A.D., Krapivkin A.I., Zavadenko N.N. A rare genetic Schuurs-Hoeijmakers syndrome (PACS1 syndrome). Epilepsy and paroxysmal conditions. 2024;16(2):120-129. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2024.183
Синдром нарушения развития нервной системы PACS1 – редкое аутосомно-доминантное генетическое заболевание, характеризующееся задержкой развития, интеллектуальным дефицитом, дисморфическими чертами, а иногда и судорогами [1–3].
Синдром PACS1 был впервые описан у двух пациентов в Нидерландах и Бельгии со схожими фенотипическими особенностями, умственной отсталостью и одинаковыми вариантами нуклеотидной последовательности в гене PACS1 [2][4]. В дальнейшем данный синдром был переименован по автору, сообщившему о нем впервые, как синдром Схюрс-Хоймакерса (Schuurs-Hoeijmakers syndrome; MIM 615009).
В настоящее время в мировой научной литературе описано 87 пациентов с нарушением развития нервной системы, обусловленным вариантами в гене PACS1. Наиболее частым был патогенный миссенс-вариант c.607C>T (р.R203W) [5]. Патогенность варианта подтверждена также сегрегационным анализом, в котором показано, что родители не были носителями, т.е. у всех больных мутация возникла de novo. Все описанные в базах данных и научных публикациях пациенты имеют характерный лицевой фенотип, задержку развития нервной системы с умственной отсталостью и психомоторными нарушениями [1–5].
В статье представлен клинический случай синдрома PACS1 у пациента с задержкой психоречевого, моторного развития и эпилепсией.
Пробанд, девочка 11 лет, наблюдалась в психоневрологическом отделении ГБУЗ «Научно-практический центр специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого Департамента здравоохранения г. Москвы» (НПЦ спец. мед. помощи детям ДЗМ) с диагнозом: «Эпилепсия фокальная, вероятно генетическая. Расходящееся содружественное альтернирующее косоглазие. Мышечная дистония».
Ведение пациентки осуществлялось сообразно принципам Хельсинкской декларации Всемирной медицинской ассоциации (Форталеза, Бразилия, 2013 г.). От родителей девочки получено информированное согласие на проведение генетического исследования (полноэкзомное секвенирование). Фото ребенка для публикации не представлено в связи с отказом родителей.
Ребенок от первой беременности, первых родов. Масса тела при рождении 3050 г, рост 50 см. Оценка по шкале Апгар 8/9. Девочка родилась доношенной, закричала сразу.
Дебют заболевания с 3 мес: на фоне инфекционного процесса и снижения температуры в стационаре зафиксирован приступ (дрожание конечностей, потеря сознания с открытыми глазами). Далее приступы повторялись каждую ночь с длительностью до 2 мин, после приступа сон продолжался. В связи со статусным характером приступов ребенок был переведен в отделение реанимации и интенсивной терапии. Пациентка выписана на терапевтической дозе леветирацетама.
Медикаментозная ремиссия длилась 1,5 года. Далее приступы возобновились, к леветирацетаму добавили вальпроевую кислоту, затем зонисамид. При повышении дозировки вальпроевой кислоты ребенок стал вялым, малоактивным, постоянно лежал. В связи с этим препарат заменили на сультиам и фенитоин. Через 5 мес частота приступов снизилась до 1 в 5–8 мес. С 3 до 5 лет на фоне проводимой противоэпилептической терапии приступы стали учащаться до 1 раза в 1–2 мес, ребенок стал тяжелее выходить из приступа.
По данным магнитно-резонансной томографии (МРТ) головного мозга, проведенной в возрасте 5 лет на аппарате с индукцией магнитного поля 3 Тл со сверхпроводящим магнитом MEXL-3010/G5 (Toshiba, Япония), зарегистрированы признаки множественных мелких врожденных аномалий: дизгенезия мозолистого тела, частичная гипоплазия червя мозжечка, гипоплазии с корковой субатрофией переднего полюса левой височной доли, гипоплазия правой верхнечелюстной пазухи. Отмечены легкая асимметрия боковых желудочков, задержка миелинизации терминальных зон.
Неврологический статус
На момент поступления ребенка в психоневрологическое отделение (возраст 11 лет) сознание ясное, судорог нет. Речь в виде простых слов. Глазные щели равные. Величина и симметрия зрачков: D=S. Менингеальный синдром не выявлен. Нистагм отсутствует. Движение глазных яблок в полном объеме. Роговичные рефлексы: D=S. Фотореакции зрачков живые. Поля зрения не изменены. Мимика симметричная. Слух не нарушен. Глотание и фонация не изменены. Нарушений чувствительности нет. Мышечная сила не изменена. Мышечный тонус повышен по типу дистонии. Сухожильные рефлексы повышены. Ходит на пальцах.
Видео-ЭЭГ-мониторинг с записью сна
Электроэнцефалография (ЭЭГ) с видеомониторингом выполнена на модульной нейродиагностической системе NicoletOne (Viasys Healthcare, США).
Основной ритм представлен нерегулярным, неустойчивым альфа-ритмом частотой 7 Гц, амплитудой 60 мкВ, с правильным зональным градиентом. Зональные различия выражены отчетливо. Медленноволновая активность диффузная, с высоким индексом, преимущественно в виде тета-колебаний, по амплитуде не превышающих фоновую ритмику. Регистрируется мультирегиональная эпилептиформная активность в виде комплеков «пик – медленная волна» амплитудой до 200 мкВ (во сне до 350 мкВ): в левых височно-теменно-затылочных, левых переднелобных, левых заднелобных, левых височных, передних вертексных, правых центрально-лобно-теменных, правых лобных отделах. Индекс представленности в бодрствовании варьируется от низких до средних значений, во сне повышается до 80–90%. Эпилептических приступов и их ЭЭГ-паттернов за время исследования не зарегистрировано (рис. 1).
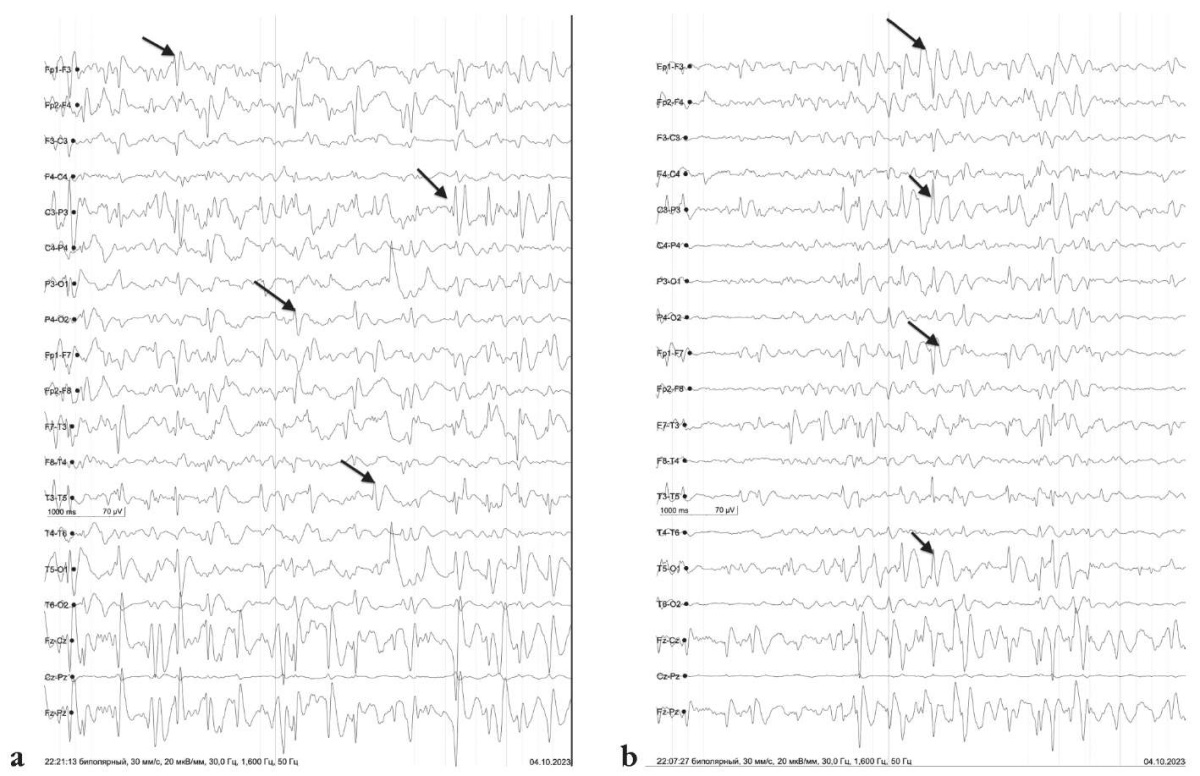
Рисунок 1. Результаты электроэнцефалографии (a, b):
стрелками показана мультирегиональная эпилептиформная активность,
представленная комплексами «пик – медленная волна» амплитудой до 200 мкВ
(во сне до 350 мкВ), в левых височно-теменно-затылочных, левых переднелобных,
левых заднелобных, левых височных, передних вертексных,
правых центрально-лобно-теменных, правых лобных отделах
Figure 1. Electroencephalography data (a, b): arrows denote multiregional epileptiform activity
in a form of peak-slow wave complexes with an amplitude of up to 200 μV
(in sleep, up to 350 μV), in the left temporo-parietal-occipital, left anterior frontal,
left posterior frontal, left temporal, anterior vertex,
right central-frontoparietal, right frontal regions
Фенотипические особенности
Рост 126 см, масса тела 27,5 кг (гипостатура). Наследственность со стороны отца не отягощена. В семье по линии матери удвоение почек с обеих сторон наблюдается в двух поколениях.
Осмысленный контакт затруднен. Команд не выполняет. Речь состоит из отдельных коротких слов. Фраз нет. Взгляд в глаза фиксирует кратковременно. Контакта избегает. Действует по собственной мотивации. Страбизм альтернирующий, расходящийся. Гипертелоризм. Короткий нос. Диспластичные ушные раковины. Макростомия. Рот приоткрыт. Язык часто вне полости рта. Аномалия прикуса. Широкие выступающие резцы на верхней челюсти. Стереотипии. Дистония. Ходит на пальцах. Деформированные стопы и ногтевые пластинки. Ограничение подвижности в голеностопных суставах. Навыки опрятности частично привиты. Порок развития почек (три почки).
Полноэкзомное секвенирование выполнено в генетической лаборатории ГБУЗ «НПЦ спец. мед. помощи детям ДЗМ»1. Геномная ДНК выделена методом лизиса клеток с последующей очисткой на стекловолоконных фильтрах (реактивы QIAamp DNA Mini Kit – Qiagen, Нидерланды), затем использована для приготовления геномных библиотек для массового параллельного секвенирования (NEBNext Ultra II – New England BioLabs, США). Из полученных библиотек методом гибридизации отобраны только те участки ДНК, которые соответствуют экзонам генов и сайтам сплайсинга (SureSelect AllExon V7, Agilent Technologies, США). Далее проведено определение их нуклеотидной последовательности на секвенаторе HiSeq 1500 (Illumina, США) (реактивы HiSeq Rapid SBS Kit v2).
Выявлен ранее описанный вариант нуклеотидной последовательности в 4-м экзоне гена PACS1 в гетерозиготном состоянии, приводящий к замене аминокислоты в 203-й позиции белка (rs398123009, chr11:6621120, c.607C>T, p.Arg203Trp). Варианты нуклеотидной последовательности в гене PACS1 в гетерозиготном состоянии описаны у пациентов с синдромом Схюрс-Хоймакерса, что предполагает аутосомно-доминантный тип наследования. Частота выявленного варианта нуклеотидной последовательности в контрольной выборке gnomAD составляет 0,0006%, в европейской популяции – 0%. Выявленный вариант нуклеотидной последовательности не зарегистрирован в российской выборке RUSeq и описан как патогенный в базе данных ClinVar (ID 39581), упоминается в научной литературе [6][7] у пациентов с синдромом Схюрс-Хоймакерса.
Поскольку выявленный вариант представлен в базе данных и научной литературе, его следует расценивать согласно критериям Американской коллегии медицинской генетики и геномики (англ. American College of Medical Genetics and Genomics, ACMG) как патогенный (сегрегационный анализ не проводился).
Синдром PACS1 включен в группу генетических нарушений клеточного транспорта (рис. 2) [8]. Хотя его клиническая характеристика хорошо описана, в научной литературе мало данных о патомолекулярных механизмах развития заболевания. Измененный вследствие мутации в гене PACS1 сортинг-белок кластера фосфофуриновой кислоты принадлежит к семейству белков – регуляторов мембранного и белкового транспорта [9].
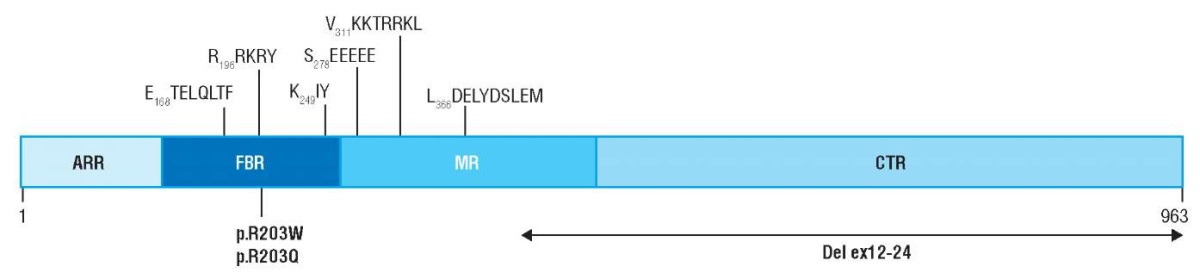
Рисунок 2. Схематическое изображение белка PACS1.
Ключевые последовательности и патологические варианты [8].
ARR (англ. atrophin-related region) – регион, связанный с атрофином;
FBR (англ. furin-binding region) – фурин-связывающий регион;
МR (англ. middle region) – средний регион;
CTR (англ. C-terminal region) – С-концевой регион
Figure 2. A schematically represented PACS1 protein.
Key sequences and pathological variants [8].
ARR – atrophin-related region; FBR – furin-binding region;
МR – middle region; CTR – C-terminal region
Первоначально белок PACS1 был обнаружен в 1998 г. [10] и описан у беспозвоночных [11] и позвоночных [12] животных. У представителей низших видов есть только один ген семейства PACS, однако в процессе эволюции с появлением позвоночных ген PACS был дуплицирован, в результате чего появились гены PACS1 и PACS2 [12].
PACS1 – многофункциональный регулятор мембранного транспорта, играющий важную роль в поддержании клеточного гомеостаза [10][12]. Первоначальной его функцией была транспортировка нескольких белков между эндосомами и сетью Гольджи [9]. Однако в последние несколько лет показаны новая роль белка как регулятора потока Ca2+ и влияние его на стабильность генома [12–14].
Ген PACS1 картирован в локусе q13.1 хромосомы 11 и включает 24 экзона [15]. Он широко экспрессируется в тканях человека [16]. Согласно базам данных BrainSpan и EvoDevo экспрессия мРНК повышена во внутриутробном периоде во время развития головного мозга и мозжечка плода с последующим снижением после рождения и незначительным увеличением в период полового созревания [15][16]. Показана важность уровня экспрессии гена в тканях тестикул в период полового созревания [17][18]. Такое специфическое распределение экспрессии гена в тканях может объяснять некоторые клинические проявления синдрома PACS1.
Все описанные в базах данных и научной литературе пациенты имеют задержку развития нервной системы с умственной отсталостью и моторными нарушениями. Клинические проявления варьируютcя от легкой до тяжелой степени выраженности [1–5]. Нарушение речи наблюдается практически у всех больных, в отличие от задержки моторного развития. Гипотония отмечается примерно у трети пациентов и с течением времени становится менее выраженной [1–5]. Тем не менее один описанный больной самостоятельно не ходил. У пациентов с патогенными вариантами в гене PACS1 отмечены и нарушения поведения различной степени выраженности. Судороги, в большинстве случаев тонико-клонические, описаны в 60% наблюдений и контролируются противоэпилептическими препаратами. По данным МРТ у пациентов с синдромом PACS1 определяли аномалии мозжечка, вентрикуломегалию, гидроцефалию или атрофию мозолистого тела [1–5].
Помимо неврологических расстройств у больных с вариантом c.607C>T (р.R203W) наблюдается характерный лицевой фенотип, который легко распознается врачом: полные и арочные брови, гипертелоризм с наклоненными вниз глазными щелями, длинные ресницы, выпуклый нос, плоский фильтр и низко посаженные большие уши [1][2].
Пациенты также могут иметь врожденные пороки сердца (дефекты перегородки) и глаз (колобома), аномалии скелета (аномальная форма черепа), крипторхизм или проблемы с питанием и другие нарушения [1][2]. Фенотип нашей пациентки с вариантом c.607C>T (р.R203W) в сравнении с больными, описанными J.H.M. Schuurs-Hoeijmakers et al. в 2016 г. [2], представлен в таблице 1.
Таблица 1. Фенотипические особенности пациентов с вариантом c.607C>T в гене PACS1
(по данным [2]) в сравнении с описанным случаем
Table 1. Phenotypic features of patients with PACS1 gene c.607C>T variant
(after [2]) compared to described case
Признак / Sign | Пациенты, описанные J.H.M. Schuurs-Hoeijmakers et al. (n=19) [2] / Patients described by J.H.M. Schuurs-Hoeijmakers et al. (n=19) [2] | Описанная нами пациентка / Current patient case |
Возраст / Age | 2–21 год / 2–21 years | 11 лет / 11 years |
Пол / Gender | 7 женщин, 12 мужчин / 7 females, 12 males | Женский / Female |
Микроцефалия / Microcephaly | 3 пациента / 3 patients | Нет / No |
Недостаточность питания / Malnutrition | 5 пациентов / 5 patients | Нет / No |
Отказ от приема пищи / Food aversion | 11 пациентов / 11 patients | Избирательна в еде / Selective in food |
Рефлюкс / Reflux | 6 пациентов / 6 patients | Нет / No |
Отставание в развитии / интеллектуальный дефицит // Developmental delay / intellectual deficit | 19 пациентов / 19 patients | Да / Yes |
Выраженное нарушение речи / Severe speech impairment | 19 пациентов / 19 patients | Да / Yes |
Расстройство аутистического спектра / Autism spectrum disorder | 6 пациентов / 6 patients | Да / Yes |
Судороги / Convulsions | 12 пациентов / 12 patients | Да / Yes |
Гипотония / Hypotension | 8 пациентов / 8 patients | Дистония. Ходьба на пальцах / Dystonia. Toe walking |
Истерика/агрессия // Hysteria/aggression | 10 пациентов / 10 patients | Да / Yes |
Структурные аномалии головного мозга / Structural brain abnormalities | 12 пациентов / 12 patients | Да / Yes |
Дисморфические черты лица / Dysmorphic facial features | 19 пациентов / 19 patients | Да / Yes |
Воронкообразная деформация грудной клетки / Funnel chest | 3 пациента / 3 patients | Нет / No |
Сколиоз / Scoliosis | 2 пациента / 2 patients | Нарушение осанки / Impaired posture |
Клинодактилия 5-го пальца / Clinodactyly of the 5th finger | 4 пациента / 4 patients | Нет / No |
Аномальная форма черепа / Abnormal skull shape | 5 пациентов / 5 patients | Нет / No |
Врожденные пороки сердца / Congenital heart disorder | 8 пациентов / 8 patients | Нет / No |
Колобома / Coloboma | 2 пациента / 2 patients | Нет / No |
Поперечная ладонная складка / Transverse palmar crease | 4 пациента / 4 patients | Бедная дерматоглифика / Poor dermatoglyphics |
Аномалия почек / Kidney abnormality | 3 пациента / 3 patients | Три почки / Three kidneys |
Аномалии глаз / Eye abnormalities | 8 пациентов / 8 patients | Страбизм / Strabismus |
Крипторхизм / Cryptorchidism | 6 пациентов / 6 patients | Нет / No |
Запор / Constipation | 9 пациентов / 9 patients | Периодически / Periodic |
Широко расставленные соски / Wide-spaced nipples | 3 пациента / 3 patients | Нет / No |
Пупочная/паховая грыжа // Umbilical/inguinal hernia | 3 пациента / 3 patients | Нет / No |
Плоскостопие / Platypodia | 4 пациента / 4 patients | Полая стопа (pes cavus) Hollow foot (pes cavus) |
Из известных на данный момент 87 случаев с вариантами в гене PACS1 наиболее частым был патогенный миссенс-вариант c.607C>T (р.R203W). Патогенность варианта подтверждена также сегрегационным анализом, в котором показано, что родители не были носителями, т.е. у всех пациентов мутация возникла de novo. У одного больного выявлен другой патогенный вариант – c.608G>A (p.R203Q) [1][2][5], однако клинические проявления у всех были схожие. В дальнейшем было идентифицировано два новых миссенс-варианта, один из которых депонирован в базе данных ClinVar (c.1574G>A; p.R525K) и его связь с синдромом PACS1 еще необходимо изучить [19]. Другой вариант выявлен у пациента с расстройствами аутистического спектра (p.R245W) без специфического фенотипа, характерного для синдрома PACS1 [20].
Помимо миссенс-вариантов в гене PACS1 описаны мультиэкзонные делеции [21]. Y. Liu et al. обнаружили у четырех членов родословной трех поколений делецию 12–24-го экзонов в гене PACS1. Однако фенотип для этого варианта был мягче, с незначительной когнитивной задержкой и нарушением речи [21]. Более того, в нескольких базах данных сообщается также о хромосомных перестройках, в которых участвует ген PACS1 (ClinVar, DECIPHER) [19][22].
При проведении фенотип-генотипических корреляций описанного нами случая с клиническими проявлениями данного синдрома у пациентов с тем же вариантом в гене PACS1, данные по которым опубликованы, клинические картины были схожи.
Таким образом, синдром PACS1 следует рассматривать у лиц со следующими клиническими проявлениями:
На сегодняшний день терапия синдрома PACS1 симптоматическая и требует мультидисциплинарного подхода со стороны специалистов разных профилей (невролог, реабилитолог, психиатр, гастроэнтеролог, кардиолог и др.). В последние годы разрабатываются таргетные подходы к лечению некоторых редких заболеваний. В этом направлении синдром PACS1 является хорошим кандидатом [23], поскольку большинство пациентов являются носителями одного и того же варианта нуклеотидной последовательности, который оказывает влияние на белок в виде усиления функции или доминантного негативного эффекта [24]. Однако высокий уровень экспрессии гена PACS1 в течение развития мозга плода может ограничить эффективность лечения [17][18].
В настоящее время существует четыре подхода: использование антисмысловых олигонуклеотидов или ингибиторов HDAC6 [25][26] находится в процессе изучения, исследование двух других зависит от прогресса в изучении 3D-структуры белка PACS1. Антисмысловые олигонуклеотиды могут специфически нацеливаться на мутантную мРНК PACS1, избегая трансляции патологического белка (рис. 3) [27]. Однако они неспособны преодолевать гематоэнцефалический барьер и требуют прямого воздействия на центральную нервную систему путем их интратекальной доставки [28]. В этом смысле интересно исследование, проведенное G. Thomas et al. [25]. Авторы предлагают конкретно воздействовать на HDAC6 путем его подавления. Применение общего или селективного ингибитора HDAC6 восстанавливает нормальный клеточный фенотип [25]. Наконец, рассматривается возможность разработки еще двух подходов, ориентированных на конкретную таргетную направленность мутированного белка PACS1. Химеры, нацеленные на протеолиз, связываются одновременно с белком-мишенью и лигазой Е3, образуя тройной комплекс, который способствует убиквитинированию интересующего белка, тем самым индуцируя протеасомальную деградацию [29]. С другой стороны, использование молекул, специфически связывающихся с мутантным PACS1, – еще один способ деградации белка. Однако развитие обеих технологий требует знания 3D-структуры белка PACS1 и различий между белками дикого и мутировавшего типов PACS1.
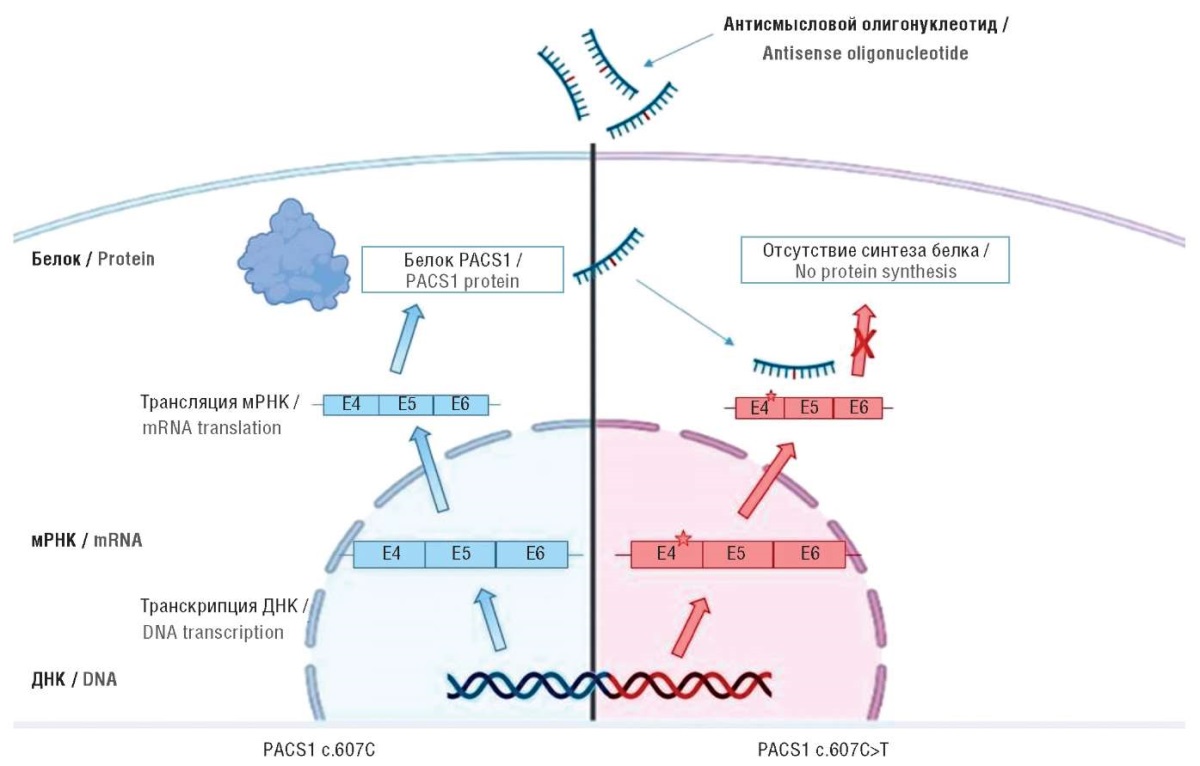
Рисунок 3. Схематическое изображение механизма терапии антисмысловыми
олигонуклеотидами для избегания трансляции мутантного белка PACS1 [27].
мРНК – матриксная рибонуклеиновая кислота;
ДНК – дезоксирибонуклеиновая кислота
Figure 3. A schematically depicted mechanism of antisense oligonucleotide therapy
for abrogating mutant PACS1 protein translation [27].
mRNA – matrix ribonucleic acid; DNA – deoxyribonucleic acid
Знание молекулярных механизмов развития синдрома PACS1 важно не только для генотип-фенотипической корреляции, но и для разработки новых терапевтических подходов в лечении, которые могли бы улучшить качество жизни пациентов. Принимая во внимание фенотипическую вариабельность и генетическую гетерогенность заболеваний, в основе которых лежит нарушение развития нервной системы, их диагностика часто вызывает затруднения.
Нами представлен клинический случай редкого генетического синдрома Схюрс-Хоймакерса (синдром PACS1), обусловленного вариантом в гене PACS1, который расширяет фенотипический спектр при данной патологии. С целью поиска причины заболевания, постановки молекулярного диагноза, а также принятия решения о тактике ведения таких пациентов необходимо выполнение генетического тестирования методом полноэкзомного или полногеномного секвенирования.
1. Научно-исследовательская работа Департамента здравоохранения г. Москвы. Тема № 123031700070-8 от 17.03.2023 г. «Персонализированный подход к диагностике, лечению и профилактике инвалидизирующих заболеваний у детей с врожденной и приобретенной патологией в многопрофильной педиатрической клинике».
1. Van Nuland A., Reddy T., Quassem F., et al. PACS1-neurodevelopmental disorder: clinical features and trial readiness. Orphanet J Rare Dis. 2021; 16 (1): 386. https://doi.org/10.1186/s13023-021-02001-1.
2. Schuurs-Hoeijmakers J.H.M., Landsverk M.L., Foulds N., et al. Clinical delineation of the PACS1-related syndrome – report on 19 patients. Am J Med Genet A. 2016; 170 (3): 670–5. https://doi.org/10.1002/ajmg.a.37476.
3. Schuurs-Hoeijmakers J.H.M., Oh E.C., Vissers L.E.L.M., et al. Recurrent de novo mutations in PACS1 cause defective cranial-neural-crest migration and define a recognizable intellectualdisability syndrome. Am J Hum Genet. 2012; 91 (6): 1122–7. https://doi.org/10.1016/j.ajhg.2012.10.013.
4. Lusk L., Smith S., Martin C., et al. PACS1 neurodevelopmental disorder. In: Adam M.P., Feldman J., Mirzaa G.M., et al. (Eds.) Gene-Reviews®. Seattle (WA): University of Washington, Seattle; 2020.
5. Arnedo M., Ascaso Á., Latorre-Pellicer A., et al. Molecular basis of the Schuurs-Hoeijmakers syndrome: what we know about the gene and the PACS-1 protein and novel therapeutic approaches. Int J Mol Sci. 2022; 23 (17): 9649. https://doi.org/10.3390/ijms23179649.
6. Tenorio-Castaño J., Morte B., Nevado J. Schuurs-Hoeijmakers syndrome (PACS1 neurodevelopmental disorder): seven novel patients and a review. Genes. 2021; 12 (5): 738. https://doi.org/10.3390/genes12050738.
7. Dutta A.K. Schuurs-Hoeijmakers syndrome in a patient from India. Case Reports Am J Med Genet A. 2019; 179 (4): 522–4. https://doi.org/10.1002/ajmg.a.61058.
8. García-Cazorla A., Oyarzábal A., Saudubray J.M., et al. Genetic disorders of cellular trafficking. Trends Genet. 2022; 38 (7): 724–51. https://doi.org/10.1016/j.tig.2022.02.012.
9. Youker R.T., Shinde U., Day R., Thomas G. At the crossroads of homoeostasis and disease: roles of the PACS proteins in membrane traffic and apoptosis. Biochem J. 2009; 421 (1): 1–15. https://doi.org/10.1042/BJ20081016.
10. Wan L., Molloy S.S., Thomas L., et al. PACS-1 defines a novel gene family of cytosolic sorting proteins required for trans-Golgi network localization. Cell. 1998; 94 (2): 205–16. https://doi.org/10.1016/s0092-8674(00)81420-8.
11. Sieburth D., Ch’ng Q., Dybbs M., et al. Systematic analysis of genes required for synapse structure and function. Nature. 2005; 436 (7050): 510–7. https://doi.org/10.1038/nature03809.
12. Thomas G., Aslan J.E., Thomas L., et al. Caught in the act – protein adaptation and the expanding roles of the PACS proteins in tissue homeostasis and disease. J Cell Sci. 2017; 130 (11): 1865–76. https://doi.org/10.1242/jcs.199463.
13. Nair-Gill E., Bonora M., Zhong X., et al. Calcium flux control by Pacs1-Wdr37 promotes lymphocyte quiescence and lymphoproliferative diseases. EMBO J. 2021; 40 (9): e104888. https://doi.org/10.15252/embj.2020104888.
14. Mani C., Tripathi K., Luan S., et al. The multifunctional protein PACS-1 is required for HDAC2- and HDAC3-dependent chromatin maturation and genomic stability. Oncogene. 2020; 39 (12): 2583–96. https://doi.org/10.1038/s41388-020-1167-x.
15. Ensembl. Gene: PACS1 ENSG00000175115. URL: https://www.ensembl.org/Homo_sapiens/Gene/Summary?db=core;g=ENSG00000175115;r=11:66070272-66244744 (дата обращения 22.01.2024).
16. GTEx Portal. PACS1. ENSG00000175115.11. URL: https://gtexportal.org/home/gene/PACS1 (дата обращения 22.01.2024).
17. Cardoso-Moreira M., Halbert J., Valloton D., et al. Gene expression across mammalian organ development. Nature. 2019; 571 (7766): 505–9. https://doi.org/10.1038/s41586-019-1338-5.
18. Evo-devo mammalian organs. Gene PACS1. URL: https://apps.kaessmannlab.org/evodevoapp/ (дата обращения 22.01.2024).
19. ClinVar. PACS1[gene]. URL: https://www.ncbi.nlm.nih.gov/clinvar/?term=PACS1%5Bgene%5D&redir=gene (дата обращения 22.01.2024).
20. Lim E.T., Uddin M., De Rubeis S., et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat Neurosci. 2017; 20 (9): 1217–24. https://doi.org/10.1038/nn.4598.
21. Liu Y., Ding H., Yan T., et al. A novel multi-exon deletion of PACS1 in a three-generation pedigree: supplements to PACS1 neurodevelopmental disorder spectrum. Front Genet. 2021; 12: 690216. https://doi.org/10.3389/fgene.2021.690216.
22. DECIPHER. PACS1 11:66070272-66244744. URL: https://www.deciphergenomics.org/gene/PACS1/overview/clinical-info (дата обращения 22.01.2024).
23. Rylaarsdam L., Reddy T., Guemez-Gamboa A. In search of a cure: PACS1 Research Foundation as a model of rare disease therapy development. Trends Genet. 2022; 38 (2): 109–12. https://doi.org/10.1016/j.tig.2021.10.010.
24. Rylaarsdam L., Guemez-Gamboa A. A gain-of-function recurrent missense variant leads to a GABAergic/glutamatergic imbalance in a forebrain organoid model of PACS1 syndrome. bioRxiv. 2022.05.13.491892. https://doi.org/10.1101/2022.05.13.491892.
25. Thomas G., Thomas L., Villar-Pazos S. Methods of treating PACS1 and PACS2 syndromes. WO 2020/018647 A1. URL: https://patents.google.com/patent/WO2020018647A1/en (дата обращения 22.01.2024).
26. PACS1 Foundation. Research Areas. URL: https://www. PACS1foundation.org/research (дата обращения 22.01.2024).
27. Rinaldi C., Wood M.J.A. Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat ReV Neurol. 2018; 14 (1): 9–21. https://doi.org/10.1038/nrneurol.2017.148.
28. Mendonça M.C.P., Kont A., Aburto M.R., et al. Advances in the design of (nano)formulations for delivery of antisense oligonucleotides and small interfering RNA: focus on the central nervous system. Mol Pharm. 2021; 18 (4): 1491–506. https://doi.org/10.1021/acs.molpharmaceut.0c01238.
29. Deshaies R.J. Protein degradation: prime time for PROTACs. Nat Chem Biol. 2015; 11 (9): 634–5. https://doi.org/10.1038/nchembio.1887.
Кожанова Татьяна Викторовна – к.м.н., ведущий научный сотрудник, врач – лабораторный генетик ГБУЗ «НПЦ специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого ДЗМ»; доцент кафедры неврологии, нейрохирургии и медицинской генетики им. академика Л.О. Бадаляна педиатрического факультета ФГАОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России.
ул. Авиаторов, д. 38, Москва 119620; ул. Островитянова, д. 1, Москва 117997
Жилина Светлана Сергеевна – к.м.н., ведущий научный сотрудник, врач-генетик ГБУЗ «НПЦ специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого ДЗМ»; доцент кафедры неврологии, нейрохирургии и медицинской генетики им. академика Л.О. Бадаляна педиатрического факультета ФГАОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России.
ул. Авиаторов, д. 38, Москва 119620; ул. Островитянова, д. 1, Москва 117997
Мещерякова Татьяна Ивановна – к.м.н., ведущий научный сотрудник, врач-генетик ГБУЗ «НПЦ специализированной медицинской помощи детям им. В.Ф. Войно-Ясенецкого ДЗМ», доцент кафедры общей и медицинской генетики медико-биологического факультета ФГАОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России.
ул. Авиаторов, д. 38, Москва 119620; ул. Островитянова, д. 1, Москва 117997
Михайлова Анна Дмитриевна – врач-невролог.
ул. Авиаторов, д. 38, Москва 119620
Крапивкин Алексей Игоревич – д.м.н., директор.
ул. Авиаторов, д. 38, Москва 119620
Заваденко Николай Николаевич – д.м.н., профессор, заведующий кафедрой неврологии, нейрохирургии и медицинской генетики им. академика Л.О. Бадаляна педиатрического факультета.
ул. Островитянова, д. 1, Москва 117997
Кожанова Т.В., Жилина С.С., Мещерякова Т.И., Михайлова А.Д., Крапивкин А.И., Заваденко Н.Н. Редкий генетический синдром Схюрс-Хоймакерса (синдром PACS1). Эпилепсия и пароксизмальные состояния. 2024;16(2):120-129. https://doi.org/10.17749/2077-8333/epi.par.con.2024.183
Kozhanova T.V., Zhilina S.S., Meshcheryakova T.I., Mikhailova A.D., Krapivkin A.I., Zavadenko N.N. A rare genetic Schuurs-Hoeijmakers syndrome (PACS1 syndrome). Epilepsy and paroxysmal conditions. 2024;16(2):120-129. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2024.183

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































