



Содержание
Перейти к:
https://doi.org/10.17749/2077-8333/epi.par.con.2024.180
Перейти к:
Представлен клинический пример пациента с синдромом Драве, вызванным мутацией в гене SCN1A. Синдром Драве – это тяжелая эпилептическая энцефалопатия, встречающаяся в раннем детском возрасте и сопровождающаяся полиморфизмом приступов, фармакорезистентным течением эпилепсии, грубыми когнитивными нарушениями. Данный случай подтверждает возможность медикаментозного контроля течения синдрома Драве. Достигнута 2-летняя ремиссия на фоне применения политерапии антиэпилептическими препаратами. В настоящее время сохраняется ремиссия на дуотерапии: топирамат в сочетании с леветирацетамом. Описанный клинический случай также демонстрирует сохранность когнитивных функций: ребенок успешно, в полном объеме осваивает общеобразовательную программу. Следует отметить, что при раннем купировании эпилептических приступов когнитивные функции не страдают.
Абусуева Б.А., Шанавазова М.Д., Аскевова М.А., Халилов В.С., Бобылова М.Ю. Случай атипичного течения тяжелой миоклонической эпилепсии младенчества (синдрома Драве). Эпилепсия и пароксизмальные состояния. 2024;16(2):130-136. https://doi.org/10.17749/2077-8333/epi.par.con.2024.180
Abusueva B.A., Shanavazova M.D., Askevova М.A., Khalilov V.S., Bobylova M.Yu. Atypical course of severe myoclonic epilepsy of infancy (Dravet syndrome). Epilepsy and paroxysmal conditions. 2024;16(2):130-136. https://doi.org/10.17749/2077-8333/epi.par.con.2024.180
У детей раннего возраста среди патологических состояний важное место занимает судорожный синдром: 10% от всех неотложных обращений за медицинской помощью [1]. Выделяют несколько основных причин судорожного синдрома: инфекционно-воспалительные заболевания центральной нервной системы, пороки развития головного мозга, гипертермический синдром, метаболические нарушения и т.д. [2][3].
Частота встречаемости фебрильных судорог в детской популяции составляет 2–4%. Простые (типичные) фебрильные судороги не сопровождаются очаговой неврологической симптоматикой, их продолжительность составляет менее 15 мин, тогда как сложные фебрильные судороги длятся более 15 мин и клинически характеризуются очаговыми симптомами [3][4].
Синдром Драве (СД) – тяжелая миоклоническая эпилепсия детства. Первыми признаками СД являются фебрильные и афебрильные генерализованные и фокальные эпилептические приступы. В типичных случаях обязательно наличие миоклонических приступов. СД характеризуется отставанием в психическом развитии и резистентностью к антиэпилептическим препаратам (АЭП) [5–7]. Согласно классификации Международной противоэпилептической лиги (англ. International League Against Epilepsy, ILAE) 2017 г. СД относится к генетическим формам эпилепсии с фебрильными приступами плюс [5]. У основной части (80%) пациентов с СД обнаруживают мутацию в гене SCN1A [8]. Частота встречаемости, по данным различных авторов, – 1 случай на 20–40 тыс. населения. Соотношение мальчиков и девочек составляет 2:1 [7].
Заболевание чаще всего начинается в первый год жизни, с пиком на 5–6-й месяц. Иногда его начало может совпадать с периодом острой респираторной вирусной инфекции (ОРВИ) или после вакцинации, однако важно понимать, что это не обязательно указывает на прямую связь между вакцинацией и патологией. Появление заболевания после вакцинации может быть связано с другими факторами, и это направление активно изучается для лучшего понимания всех аспектов данной проблемы.
В течении заболевания можно выделить три стадии. Первая – до 1 года, стадия дебюта, период клонических приступов, в основном фебрильных. Вторая – катастрофическая стадия в возрасте 1–4 года. Наблюдаются фебрильные и афебрильные приступы (т.е. возникающие на фоне соматического благополучия ребенка). К ним относятся фокальные моторные приступы, миоклонические, атипичные абсансы, альтернирующие гемиклонии, тонико-клонические приступы. Однако ключевым звеном в клинической картине является эпилептический миоклонус (активный и негативный). Все приступы имеют тенденцию к статусному течению (30 мин и более), резистентны к АЭП-терапии [6]. С момента учащения частоты приступов отмечается выраженная задержка психического развития ребенка с дальнейшим регрессом ранее приобретенных психоречевых навыков, в то время как моторное развитие заметно не нарушается [11]. Третья стадия (стабилизация) наступает после 4–5 лет, частота приступов снижается, превалируют грубые когнитивные и неврологические нарушения [5][6].
При электроэнцефалографическом (ЭЭГ) исследовании отмечается замедление основной активности, а также доминирование тета-ритма с частотой 4–5 Гц. В межприступном периоде в 70% случаев регистрируется диффузная и региональная эпилептиформная активность. У 50% пациентов иктальная ЭЭГ характеризуется фотосенситивностью, иногда возможна аутоиндукция приступов ярким светом. Эпилептиформная активность может провоцироваться закрыванием глаз, фотосенситивной реакцией и имеет тенденцию к нарастанию во время сна. Следует отметить, что с момента начала заболевания до установления диагноза проходит не менее 1–2 лет [6].
Основными критериями диагностики СД являются [12]:
В рамках дифференциальной диагностики СД следует рассматривать эволюционную и эпилептическую энцефалопатию (англ. developmental and epileptic encephalopathy, DEE), связанную с мутацией в гене PCDH19. Этот синдром сопровождается эпилептическими приступами, интеллектуальной недостаточностью, но ограничен женским полом. В клинической картине у некоторых пациентов с указанными заболеваниями наблюдаются фебрильные и афебрильные приступы в младенческом возрасте, серийное и статусное течение пароксизмов во время лихорадки, регресс развития после дебюта [13]. Вследствие того что эти два заболевания имеют схожую клиническую картину, необходимо исследование на мутацию в гене PCDH19 у пациентов с клиническими признаками СД и отрицательным генетическим анализом на мутацию SCN1A [9].
Эпилепсия при СД характеризуется фармакорезистентным течением. Важно не допускать подъема температуры тела [14]. Препаратами выбора для лечения СД являются вальпроаты [15], они особенно эффективны при атипичных абсансах, эпилептическом миоклонусе. Вальпроаты применяют в дозе 30–80 мг/кг/сут. Отмечается особая эффективность топирамата при СД: он эффективен при генерализованных судорожных приступах и гемиконвульсиях с альтернирующей локализацией. Препарат назначают начиная с 12,5 мг/сут с постепенным повышением по 12,5 мг в неделю до дозы 50–200 мг/сут в 2 приема [15]. Блокаторы натриевых каналов (карбамазепин, ламотриджин, фенитоин, вигабатрин) при СД противопоказаны, т.к. имеют высокий риск аггравации приступов, в частности миоклонических [16][17]. При неэффективности монотерапии применяются комбинации АЭП, из которых самым предпочтительным является сочетание топирамата и вальпроата. Другая эффективная комбинация – топирамат и леветирацетам или вальпроаты-сукцинимиды. Этосуксимид назначается в дозировке 250–750 мг/сут в 2–3 приема [15].
В работе М.Ю. Бобыловой [6] приводится опыт применения перампанела при СД на примере двух случаев. Автор подчеркивает высокую эффективность перампанела в качестве дополнительной терапии: у одной пациентки медикаментозная ремиссия была достигнута на 3 года в отношении всех приступов, а у второго пациента афебрильные приступы купировались, отмечались только фебрильные приступы на фоне ОРВИ. Применение других методов лечения (кортикостероиды, иммуноглобулины, кетогенная диета, стимуляция блуждающего нерва) показали низкую эффективность при СД [6].
С целью борьбы с фотосенситивностью и аутоиндукцией пароксизмов рекомендуется ношение очков с голубыми стеклами. Очень важно не допускать повышения температуры тела, а при гипертермии необходимо срочное применение жаропонижающих средств. Следует исключать горячие ванны и перегревание [6][18].
Заболевание имеет неблагоприятный прогноз. При СД инвалидизация чаще всего глубокая. Ремиссия наступает в единичных случаях, как правило, на непродолжительное время [15][19]. Следовательно, основными целями лечения являются снижение частоты приступов, профилактика серийного и статусного течения, улучшение когнитивных функций. СД – это эволюционная и эпилептическая энцефалопатия, т.е. помимо эпилепсии в клинической картине отмечаются интеллектуальные расстройства, в значительной степени приводящие к социальной дезадаптации пациента [11]. В подростковом возрасте приступы становятся реже, однако в неврологическом статусе превалируют экстрапирамидные расстройства, выраженные когнитивные нарушения.
Летальность в младенчестве составляет около 20% [6][7]. У взрослых пациентов одной из частых причин смерти во сне является синдром внезапной смерти при эпилепсии (англ. sudden unexpected death in epilepsy, SUDEP). Второе место по частоте занимает эпилептический статус [20].
Представляем клиническое наблюдение ребенка с тяжелой эпилепсией младенчества (синдромом Драве) – пациентки С. 2015 г.р.
Ребенок от первой беременности, роды протекали без патологии. После рождения закричала сразу, по шкале Апгар 8–9 баллов. До 1 года развивалась соответственно возрасту. С рождения обращало на себя внимание нарушение терморегуляции, температура тела повышалась при нахождении ребенка в теплой одежде, в жаркую погоду. Девочка плохо переносила солнце, относительно хорошо – горячую ванну и душ. Семейный анамнез по эпилепсии не отягощен.
Первый приступ случился в 6 мес во сне – продолжительностью 10–15 мин с миоклоническими подергиваниями на фоне гипертермии. Далее приступы повторялись в 10 и 12 мес, также на фоне лихорадки, но уже со статусным течением 40–45 мин и развитием постиктального паралича Тодда, купировались реланиумом. Позже возникали как фебрильные, так и афебрильные приступы.
После 1 года стал отмечаться неврологический дефицит в виде гипотонии, шаткой походки, задержки психоречевого развития. На ЭЭГ в 1 год зарегистрирована эпилептиформная активность – в правой височно-затылочной и левой лобно-центральной областях наблюдались одиночные комплексы «острая – медленная волна».
Полноэкзомное секвенирование проводили на платформе NextSeq 500 (lllumina, США) в лаборатории «Геномед» (Россия). По результатам секвенирования ДНК (панель «Наследственные эпилепсии») обнаружена гетерозиготная мутация в экзоне 6 гена SCN1A, приводящая к делеции аминокислоты в 318-й позиции.
В возрасте 10 мес была предпринята попытка терапии вальпроевой кислотой в каплях для приема внутрь, затем в микрогранулах – без эффекта, приступы повторялись. Положительный эффект проявился при введении топирамата в дозе 25 мг 2 раза в день в сочетании с вальпроатом в дозе 100 мг 2 раза в день.
В 1 год 6 мес в связи с имеющимися жалобами на шаткость походки, частые падения во время ходьбы, снижение мышечного тонуса, задержку речи, снижение концентрации внимания, несформированность игровой деятельности, эмоциональную лабильность для пациентки была разработана реабилитационная программа. Она включала занятия с логопедом, лечебную физкультуру, физиотерапию (тепловые процедуры, электрофорез, парафинотерапию на дистальные отделы конечностей), уроки плавания в бассейне.
В возрасте 2 лет появились приступы в виде замираний, а при ультразвуковом исследовании (УЗИ) выявлены гепато- и спленомегалия, а также определялись изменения биохимических показателей крови, расцененные как нежелательные явления. Поэтому пациентку перевели с вальпроата на леветирацетам в дозе 180 мг 2 раза в день, повысили дозировку топирамата до 50 мг 2 раза в день. В момент повышения дозы отмечалась редукция приступов на несколько недель.
В 2 года 4 мес произошло ухудшение – три статусных приступа за 1 мес. Два приступа длительностью 40–45 мин, третий приступ фебрильный, который не удавалось купировать более 2 ч, поэтому бригадой скорой помощи пациентка была доставлена в диагностическое отделение ГБУ Республики Дагестан «Детская республиканская клиническая больница им. Н.М. Кураева».
Состояние было расценено как тяжелое. По завершении приступа у ребенка отмечались тошнота и рвота, положительная менингеальная симптоматика (ригидность затылочных мышц – два пальца, симптом Кернига 140–150 градусов). В связи с этим пациентка была пунктирована, но в спинномозговой жидкости значимых изменений не обнаружено. После проведения люмбальной пункции состояние больной улучшилось, симптоматика регрессировала. Принято решение нарастить дозу топирамата до 75 мг 2 раза в день.
В течение последующих 3 лет вновь случались приступы при лихорадке на фоне ОРВИ и около двух-трех на фоне соматического здоровья. Весь период наблюдения проводился контроль УЗИ и биохимических показателей функции печени и почек: после замены вальпроата на леветирацетам все было в пределах нормы.
В возрасте 6 лет на фоне приема АЭП наступила ремиссия по приступам. Девочка продолжает посещать занятия с логопедом-дефектологом, по нейросенсорике, нейродинамической гимнастике, а также уроки плавания в бассейне. Курсами проводится массаж с исключением шейно-воротниковой зоны и головы, выполняются микрополяризация и тепловые процедуры.
С наступлением ремиссии отмечен выраженный прогресс в психомоторном и речевом развитии на фоне применения методов физической реабилитации, занятий с психологом, логопедом, сенсорной интеграции, нейрогимнастики. Несмотря на тяжелый инвалидизирующий диагноз, к 5 годам пациентка овладела многими новыми навыками и знаниями, пользуется интерактивными игрушками, собирает сложные конструкции. К 8 годам пошла в школу, успешно, в полном объеме осваивает общеобразовательную программу, навыки опрятности и самообслуживания полностью сформированы, помогает в уходе за младшим братом.
Данные видео-ЭЭГ-мониторинга от 2018 г., где регистрируется региональная эпилептиформная активность на затылочно-височном отведении слева, представлены на рисунке 1. В 2023 г. основной ритм в пределах возрастной нормы и эпилептиформная активность отсутствует.
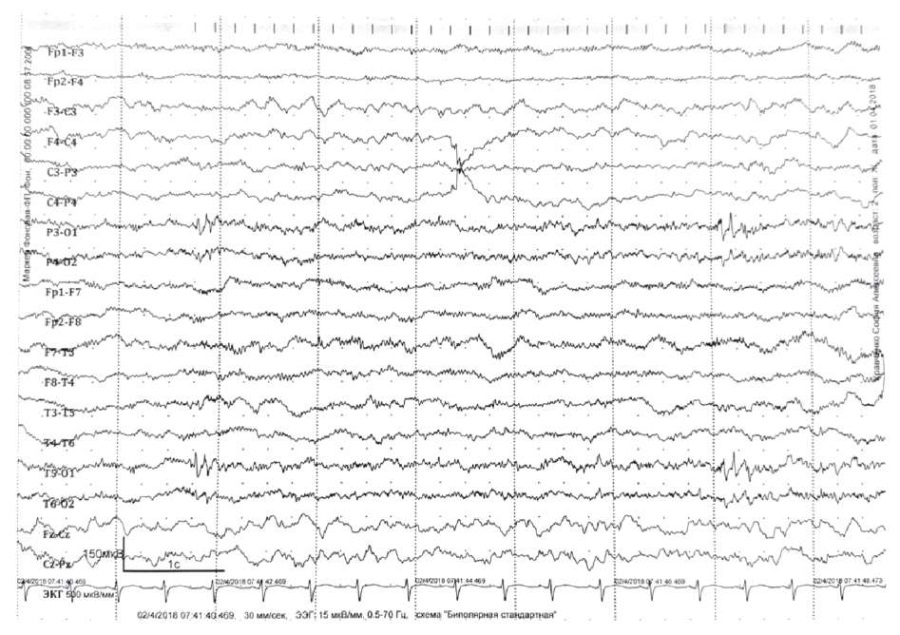
Рисунок 1. Электроэнцефалограмма пациентки от 2018 г.
Региональная эпилептиформная активность – разряд полиспайков
в левой задне-височной и теменно-центральной зонах амплитудой до 120 мкВ
Figure 1. The patient’s electroencephalogram dated 2018.
Regional epileptiform activity – polyspike discharge
in the left posterior-temporal and parietal-central zones, amplitude up to 120 μV
Синдром Драве является труднокурабельным эпилептическим синдромом [14], который дебютирует на первом году жизни у ребенка с нормальным анамнезом и психомоторным развитием. В дебюте приступы могут быть короткими и продолжительными, как правило, фебрильно провоцируемыми [5][7][9]. В представленном нами случае у пациентки приступы дебютировали в 6 мес на фоне гипертермии.
Фебрильные приступы возникают даже при невысокой температуре, имеют фокальный характер, в основном это альтернирующие гемиклонические приступы с трансформацией в эпилептический статус [10]. Статусное течение наблюдалось и у нашей пациентки. Сохранявшиеся приступы характеризовались полиморфизмом – билатеральные тонико-клонические приступы, серийные миоклонии, фокальные приступы с последующей вторичной генерализацией и резистентностью к АЭП-терапии.
По данным Е.Д. Белоусовой и др., у пациентов с СД развивается пирамидная недостаточность в виде гиперрефлексии, диффузной мышечной гипотонии, моторная неловкость, атаксия [9]. После 7 лет присоединяются экстрапирамидные нарушения (гипокинезия, тремор, повышение мышечного тонуса по пластическому типу) [5], в пубертатном возрасте возникают ортопедические осложнения (кифосколиоз, плоскостопие) [6].
В нашем случае ребенок развивался до 1 года соответственно возрасту, что не противоречит данным литературы. После 1 года у пациентки отмечен неврологический дефицит в виде гипотонии, шаткой походки, задержки психоречевого развития. С момента появления в 1 год 6 мес очаговой неврологической симптоматики девочка получала необходимый объем реабилитационного лечения с учетом показаний и противопоказаний. К тому же удалось купировать эпилептические приступы. По нашему мнению, это дало возможность компенсировать задержку развития.
В представленном случае у пациентки в 1 год на ЭЭГ регистрировалась региональная эпилептиформная активность, которая коррелировала с ухудшением клинической картины. Видео-ЭЭГ-мониторинг в возрасте 8 лет демонстрирует возрастную норму основного ритма и отсутствие эпилептиформной активности. По литературным данным, у пациентов с СД при ЭЭГ-исследовании диагностируют замедление основной активности с доминированием тета-ритма частотой 4–5 Гц. В межприступном периоде в 70% случаев регистрируется диффузная и региональная эпилептиформная активность с нарастанием во время сна, характерна фотосенситивность [6].
Как уже было отмечено, при генерализованных судорожных приступах и гемиконвульсиях с альтернирующей локализацией при СД эффективен топирамат, который назначают начиная с 12,5 мг/сут с постепенным повышением дозы по 12,5 мг в неделю до дозы 50–200 мг/сут в 2 приема [15].
В нашем клиническом случае положительный результат отмечен именно при применении топирамата. Также девочка получала вальпроат, на фоне которого наблюдалось улучшение по приступам, но развились нежелательные явления. Проведена постепенная отмена вальпроата и переход на леветирацетам. В 6 лет на фоне дуотерапии АЭП (леветирацетам и топирамат) была достигнута клиническая ремиссия по приступам. К моменту написания статьи регистрировалась полная клинико-электроэнцефалографическая ремиссия, при этом клиническая ремиссия сохраняется на протяжении 30 мес.
Представленный клинический случай демонстрирует целесообразность назначения политерапии АЭП при СД, цель которой состоит в максимально быстром достижении и поддержании ремиссии и, как следствие, улучшении исхода заболевания, а также в уменьшении степени выраженности когнитивных нарушений. Примененные реабилитационные методы показали свою безопасность и эффективность.
Данный пример доказывает возможность благоприятного исхода при СД в случае успешного и быстрого купирования эпилептических приступов, а также при проведении реабилитационных и абилитационных мероприятий, при комплаентности пациента, его законного представителя и врача. Это ставит под сомнение однозначность неблагоприятного исхода при такой патологии. Наше наблюдение уникально в своем роде, в мировой литературе встречаются единичные случаи подобного течения.
1. Карлов В.А. Пошаговая терапия эпилептического статуса. Общие рекомендации по дозам и схемам введения. М.; 2009: 3.
2. Долинина А.Ф. Клиническая и генетическая характеристики синдрома Драве. Педиатрический вестник Южного Урала. 2017; 2: 37–40.
3. Холин А.А., Ильина Е.С., Колпакчи Л.М. и др. Злокачественные мигрирующие парциальные приступы младенчества. Клиническое наблюдение 6 случаев. Русский журнал детской неврологии. 2007; 2 (2): 25–38.
4. Hernandez M., Pedraza M., Mesa T., Troncoso M. Complex febrile seizures or Dravet syndrome? Description of 3 case reports. Rev Chil Pediatr. 2014; 85 (5): 588–93 (на исп. яз.). https://doi.org/10.4067S0370-41062014000500010.
5. Бобылова М.Ю. Резистентные формы эпилепсии, ассоциированные с фебрильно-провоцируемыми приступами (cиндромы Драве, DESC, HHE) (лекция). Русский журнал детской неврологии. 2012; 7 (4): 31–40.
6. Бобылова М.Ю. Опыт применения перампанела при синдроме Драве на примере двух случаев. Вестник эпилептологии. 2020; 1: 20–5.
7. Хузина Г.Р., Закирова Д.Р., Тухбатуллина Э.Л., Иксанова Е.Н. Синдром Драве. Вестник современной клинической медицины. 2013; 6 (Прил. 1): 49–52.
8. Rosander C., Halböök T. Dravet syndrome in Sweden: a population-based study. Dev Med Child Neurol. 2015; 57 (7): 628–33. https://doi.org/10.1111/dmcn.12709.
9. Белоусова Е.Д., Шарков А.А. Трудности в диагностике, прогнозе и лечении генетических эпилептических энцефалопатий: взгляд невролога. Журнал неврологии и психиатрии им. С.С. Корсакова. 2019; 119 (11-2): 34–40. https://doi.org/10.17116/jnevro201911911234.
10. Мухин К.Ю., Петрухин А.С., Холин А.А. Эпилептические энцефалопатии и схожие синдромы у детей. М.: Арт-Сервис Лтд; 2011: 157–74.
11. Домбровская Е.А., Пуголовкин К.А. Синдром Драве – тяжелая миоклоническая эпилепсия младенчества: опыт достижения 2-летней ремиссии у ребенка 2 лет 7 месяцев. Педиатрия. 2016; 95 (5): 168–71.
12. Guerrini R. Dravet syndrome: the main isues. Eur J Paediatr Neurol. 2012; 16 (Suppl. 1): S1–4. https://doi.org/10.1016/j.ejpn.2012.04.006.
13. Мухин К.Ю., Пылаева О.А., Долинина А.Ф. и др. Эпилепсия, вызванная мутацией гена PCDH 19: обзор литературы и собственные наблюдения. Русский журнал детской неврологии. 2016; 11 (2): 26–32. https://doi.org/10.17650/2073-8803-2016-11-2-26-32.
14. Abril Jaramillo J., Estevez Maria J.C., Girón Úbeda J.M., et al. Effectiveness and safety of perampanel as early add-on treatment in patients with epilepsy and focal seizures in the routine clinical practice: Spain prospective study (PERADON). Epilepsy Behav. 2020; 102: 106655. https://doi.org/10.1016/j.yebeh.2019.106655.
15. Мухин К.Ю., Бобылова М.Ю., Чадаев В.А., Петрухин А.С. Эпилептические синдромы. Диагностика и терапия. 4-e изд. М.: Бином; 2018: 608 с.
16. Wallace A., Wirrell E., Kenney-Jung D.L. Pharmacotherapy for Dravet syndrome. Paediatr Drugs. 2016; 18 (3): 197–208. https://doi.org/10.1007/s40272-016-0171-7.
17. Wu Y.W., Sullivan J., McDaniel S.S., et al. Incidence of Dravet syndrome in a US population. Pediatrics. 2015; 136 (5): e1310–5. https://doi.org/10.1542/peds.2015-1807.
18. Brunklaus A., Zuberi S.M. Dravet syndrome – from epileptic encephalopathy to channelopathy. Epilepsia. 2014; 55 (7): 979–84. https://doi.org/10.1111/epi.12652.
19. Kang J.Q., Macdonald R.L. Molecular pathogenic basis for GABRG2 mutations associated with a spectrum of epilepsy syndromes, from generalized absence epilepsy to Dravet syndrome. JAMA Neurol. 2016; 73 (8): 1009–16. https://doi.org/10.1001/jamaneurol.2016.0449.
20. Cooper M.S., Mcintosh A., Crompton D.E., et al. Mortality in Dravet syndrome. Epilepsy Res. 2016; 128: 43–7. https://doi.org/10.1016/j.eplepsyres.2016.10.006.
Абусуева Бурлият Абусуевна – к.м.н., доцент, заведующая кафедрой нервных болезней, медицинской генетики и нейрохирургии.
пл. Ленина, д. 1, Махачкала 367000, Республика Дагестан
Шанавазова Мукминат Джабраиловна – ассистент кафедры нервных болезней, медицинской генетики и нейрохирургии.
пл. Ленина, д. 1, Махачкала 367000, Республика Дагестан
Аскевова Мариям Абдулмажитовна – ассистент кафедры нервных болезней, медицинской генетики и нейрохирургии.
пл. Ленина, д. 1, Махачкала 367000, Республика Дагестан
Халилов Варис Садрутдинович – к.м.н., доцент кафедры неврологии, нейрохирургии и медицинской генетики им. академика Л.О. Бадаляна педиатрического факультета ФГАОУ ВО «РНИМУ им. Н.И. Пирогова» Минздрава России; врач-рентгенолог отделения магнитно-резонансной томографии ФГБУ «ФНКЦ детей и подростков ФМБА».
ул. Островитянова, д. 1, Москва 117997; ул. Москворечье, д. 20, Москва 115409
Бобылова Мария Юрьевна – к.м.н., доцент, врач – невролог-эпилептолог.
ул. Нагорная, д. 8, Москва 108842
Абусуева Б.А., Шанавазова М.Д., Аскевова М.А., Халилов В.С., Бобылова М.Ю. Случай атипичного течения тяжелой миоклонической эпилепсии младенчества (синдрома Драве). Эпилепсия и пароксизмальные состояния. 2024;16(2):130-136. https://doi.org/10.17749/2077-8333/epi.par.con.2024.180
Abusueva B.A., Shanavazova M.D., Askevova М.A., Khalilov V.S., Bobylova M.Yu. Atypical course of severe myoclonic epilepsy of infancy (Dravet syndrome). Epilepsy and paroxysmal conditions. 2024;16(2):130-136. https://doi.org/10.17749/2077-8333/epi.par.con.2024.180

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































