


Содержание
Перейти к:
 Е. Е. Тимечко,
О. С. Шилкина,
Н. В. Орешкова,
В. О. Кобаненко,
Е. А. Осипова,
Н. А. Шнайдер,
Д. В. Дмитренко
Е. Е. Тимечко,
О. С. Шилкина,
Н. В. Орешкова,
В. О. Кобаненко,
Е. А. Осипова,
Н. А. Шнайдер,
Д. В. Дмитренко https://doi.org/10.17749/2077-8333/epi.par.con.2022.119
Перейти к:
Актуальность. Ювенильная миоклоническая эпилепсия (ЮМЭ) является наиболее распространенным типом идиопатической генерализованной эпилепсии с дебютом в подростковом и взрослом возрасте. При медико-генетическом консультировании у пробандов с ЮМЭ нередко выявляется отягощенная наследственность по эпилепсии. Однако конкретные генетические варианты предрасположенности к ЮМЭ остаются неубедительными. Использование современных методов генетического анализа, в частности проведение полноэкзомного и полногеномного секвенирования, позволяет обнаружить, подтвердить и упрочнить ассоциативную связь определенного патологического фенотипа с наличием того или иного патогенного варианта в ряде генов.
Цель: анализ результатов полноэкзомного секвенирования у пациентов с ЮМЭ и поиск ассоциативных связей с заболеванием.
Материал и методы. В исследование включены 7 пациентов с установленным диагнозом ЮМЭ и 1 ребенок пробанда без клинических признаков эпилепсии. Полноэкзомное секвенирование проведено с использованием аппарата MiSeq (Illumina, США), биоинформатический анализ осуществлен на платформе Genomenal (Novel Software Systems, Россия).
Результаты. Выявлено гетерозиготное носительство патогенных вариантов в генах рецессивных заболеваний: SACS, AHI1, CEP164, ANO10, RMND1, POMGNT1, FLG, ACTB. При анализе обнаруженных генетических вариантов у обследованных пациентов ассоциаций с клинической картиной заболевания не установлено. Отмечены гетерозиготные миссенсмутации в генах CLCN2, EFHC1, JRK, ME2 и frameshift-мутация в гене CACNB4.
Заключение. В последние годы значительные усилия направлены на идентификацию генов предрасположенности к ЮМЭ. В проведенном нами исследовании моногенных и/или полигенных патогенных вариантов у пациентов с ЮМЭ и ребенка пробанда с ЮМЭ не выявлено. Высокая генетическая гетерогенность заболевания может объяснить многочисленные безуспешные попытки найти гены предрасположенности к ЮМЭ. Необходимы дальнейшие исследования для подтверждения вариантов, ассоциированных с развитием ЮМЭ. Достижения в области геномных технологий могут расширить наше понимание генетики данной патологии.
Тимечко Е.Е., Шилкина О.С., Орешкова Н.В., Кобаненко В.О., Осипова Е.А., Шнайдер Н.А., Дмитренко Д.В. Полноэкзомное секвенирование пациентов с юношеской миоклонической эпилепсией. Эпилепсия и пароксизмальные состояния. 2022;14(3):254-266. https://doi.org/10.17749/2077-8333/epi.par.con.2022.119
Timechko E.E., Shilkina O.S., Oreshkova N.V., Kobanenko V.O., Osipova E.A., Shnayder N.A., Dmitrenko D.V. Whole-exome sequencing of patients with juvenile myoclonic epilepsy. Epilepsy and paroxysmal conditions. 2022;14(3):254-266. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2022.119
Юношеская миоклоническая эпилепсия (ЮМЭ) является наиболее распространенной формой эпилепсии, на ее долю приходится около 9,3% всех эпилепсий [1]. ЮМЭ характеризуется наличием абсансов, миоклонических приступов и генерализованных тонико-клонических приступов (ГТКП), обычно возникающих после пробуждения. Члены семьи пациента с ЮМЭ часто также страдают эпилепсией с вариабельным фенотипом [2].
Согласно классификации эпилепсии Международной Противоэпилептической Лиги (англ. International League Against Epilepsy, ILAE) 2017 г. ЮМЭ относится к генетическим генерализованным эпилепсиям (ГГЭ) [3]. Однако в 2021 г. рабочая группа по нозологии и определениям ILAE в драфт-версии публикации выделила четыре формы ГГЭ в отдельную группу идиопатической генерализованной эпилепсии (ИГЭ) [4].
ИГЭ – группа зависящих от возраста расстройств с характерными отличительными электро-клиническими особенностями и известной или предполагаемой генетической этиологией [4]. ИГЭ характеризуются наличием абсансов, миоклонических приступов и ГТКП [5] с дебютом в детском или юношеском возрасте и являются одной из наиболее распространенных форм эпилепсии, составляя до 1/3 от всех форм заболевания [6]. К ИГЭ относят следующие формы: детская абсансная эпилепсия (ДАЭ), ЮМЭ, юношеская абсансная эпилепсия (ЮАЭ) и эпилепсия с ГТКП [7].
Клинические близнецовые и семейные исследования показывают, что ИГЭ могут быть генетически обусловлены [2][8]. Монозиготные близнецы высококонкордантны, со 100% конкордантностью по электроэнцефалографическому признаку: генерализованная спайкволновая активность, 70% совпадений эпилептических приступов [9][10]. Долгое время ученые считали, что в основе ИГЭ лежат сложные механизмы наследования, подразумевая возможную полигенную природу с наличием либо отсутствием влияния факторов окружающей среды [4].
Однако, несмотря на клиническое подтверждение наследственного характера ИГЭ, патогенные варианты, ассоциированные с развитием данных форм заболевания, до сих пор недостаточно изучены [4].
В небольшой части случаев выявлены моногенные причины ИГЭ, включающие гены, кодирующие субъединицы рецептора гамма-аминомасляной кислоты (ГАМК) (например, GABRG2, GABRA1) [11][12], и ген, кодирующий транспортер глюкозы 1 (SLC2A1) [13]. Описаны как унаследованные от родителей, так и de novo мутации. В случае наследуемых мутаций возможна неполная пенетрантность патогенного варианта. У большого числа больных ИГЭ родословная не отягощена по эпилепсии, что объясняется либо мутацией de novo, либо сложным наследованием [14].
Кроме моногенных вариантов у 3% пациентов с ИГЭ встречаются варианты числа копий: микроделеции и микродупликации [15][16]. В настоящее время считается, что они вносят свой вклад в этиологию, но не являются причиной заболевания. Данные генетические варианты могут быть семейными или возникать de novo и существенно увеличивать риск ИГЭ [17].
Клинические близнецовые исследования показали генетический вклад в патогенез ЮМЭ [4]. В ряде исследований сообщалось о носительстве патогенных вариантов у больных ЮМЭ в генах CACNB4, GABRA1, GABRD и EFHC1 [18–20].
Однако генетическое тестирование не является частью текущей рутинной диагностической практики как при ЮМЭ, так и при других формах ИГЭ. Поиск этиологических причин развития ЮМЭ продолжается в настоящее время.
Цель – анализ результатов полноэкзомного секвенирования у пациентов с ЮМЭ и поиск ассоциативных связей с заболеванием.
Для генетического исследования на базе Неврологического центра Университетской клиники ФГБОУ ВО «Красноярский государственный медицинский университет им. профессора В.Ф. Войно-Ясенецкого» Минздрава России были последовательно набраны 7 пациентов с подтвержденным диагнозом ЮМЭ согласно критериям ILAE и 1 ребенок пробанда без клинических признаков заболевания на момент проведения исследования (табл. 1).
Таблица 1. Клинические характеристики обследованных пациентов
Table 1. Clinical characteristics of the patients examined

Примечание. ГТКП – генерализованные тонико-клонические приступы; ЮМЭ – юношеская миоклоническая эпилепсия.
Note. GTCS – generalized tonic-clonic seizures; JME – juvenile myoclonic epilepsy
Исследование было выполнено в соответствии со стандартами надлежащей клинической практики и принципами Хельсинкской декларации Всемирной медицинской ассоциации 2013 г., одобрено локальным этическим комитетом. Все участники подписали информированное согласие.
Геномная ДНК была выделена из периферической крови пациентов с использованием коммерческого набора QIAmp DNA MiniKiT (Helicon, Россия) по стандартному для него протоколу на оборудовании центра коллективного пользования «Молекулярные и клеточные технологии» ФГБОУ ВО «Красноярский государственный медицинский университет им. профессора В.Ф. Войно-Ясенецкого» Минздрава России. Экзом пациентов получен обогащением образцов ДНК с помощью коммерческого набора Clinical exome solution v2 (Sophia Genetics, Швейцария) по стандартному протоколу. Обогащенные библиотеки далее секвенировались с использованием платформы MiSeq Illumina: MiSeq Reagent Kit v3 (2×300bp) (Illumina, США) в лаборатории лесной геномики Института фундаментальной биологии и биотехнологии ФГАОУ ВО «Сибирский федеральный университет». Длина полученных парных прочтений составляла не менее 250 пар оснований.
Парные прочтения, полученные после секвенирования, были предоставлены в формате FASTq. Для их анализа применяли специализированную платформу Genomenal: NGS Wizard (Novel Software Systems, Россия). Фильтрацию проводили с использованием двух панелей генов:
1) панель «Наследственные эпилепсии», включающая наиболее полный список ассоциированных с эпилепсией генов (всего 977), разделенных на четыре категории в зависимости от проявления заболевания в фенотипе:
84 гена, ассоциированные только с эпилепсией или синдромами эпилепсии в качестве основного симптома,
73 гена, ассоциированные с врожденными пороками развития головного мозга и эпилепсией,
536 генов, связанных с грубыми нарушениями физического развития или другими системными отклонениями, сопровождающимися эпилептическими приступами,
284 гена-кандидата, требующие дальнейшей проверки [21];
2) панель «ЮМЭ», включающая гены, ассоциированные с развитием ЮМЭ: BRD2, CACNB4, CHRNA4, CLCN2, CX36, EFHC1, GABRA1, GABRD, JRK, ME2 GRM4, CHRM3 [18].
Обнаруженные варианты фильтровались с помощью сервиса для автоматической обработки геномных данных NGS Wizard на платформе Genomenal (Novel Software Systems, Россия), обращающейся к базам данных ClinVar [22], nomAD [23], ExAC [24], OMIM [25], Ensembl [26], NCBI [27], UniProt [28]. Варианты были отсортированы по значению эффекта. Варианты со значением High принимались за патогенные. Это значение получали варианты, приводящие к таким генетическим перестройкам, как frameshift, start lost, splice donor, splice acceptor и stop gained. Найденные варианты были классифицированы с использованием рекомендаций для интерпретации данных, полученных методами массового параллельного секвенирования [29].
При анализе данных, полученных с использованием общей панели генов эпилепсии, было идентифицировано 59 вариантов, среди них 42 с невыясненной патогенностью, 2 вероятно патогенных и 8 патогенных (табл. 2).
Таблица 2. Патогенные, вероятно патогенные, доброкачественные и вероятно доброкачественные варианты у пациентов с юношеской миоклонической эпилепсией (панель «Наследственные эпилепсии») Table 2. Pathogenic, likely pathogenic, benign, and potentially benign variants in patients with juvenile myoclonic epilepsy (Hereditary Epilepsy Panel)
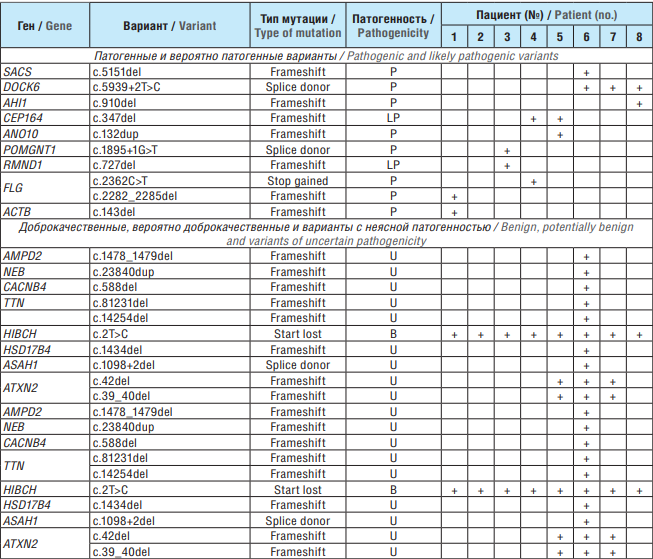
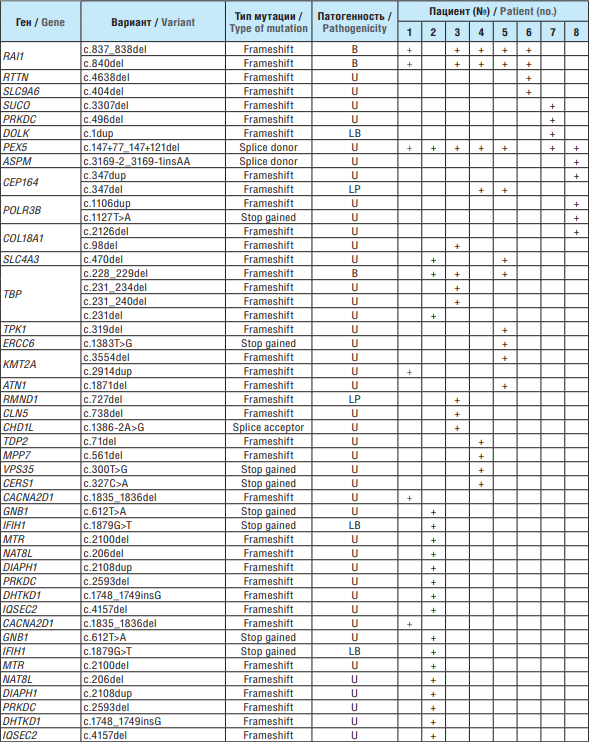
Примечание. P (англ. pathogenic) – патогенная; LP (англ. likely pathogenic) – вероятно патогенная; U (англ. uncertain) – неясной патогенности; B (англ. benign) – доброкачественная; LB (англ. likely benign) – вероятно доброкачественная.
Note. P – pathogenic; LP – likely pathogenic; U – uncertain; B – benign; LB – likely benign.
Выявлено гетерозиготное носительство патогенных вариантов в генах рецессивных заболеваний: SACS, AHI1, CEP164, ANO10, RMND1, POMGNT1, FLG, ACTB (см. табл. 1). Изменения в данных генах могут быть ассоциированы с патологическими фенотипами, для которых наличие эпилептического приступа не является обязательным симптомом. При анализе найденных генетических вариантов у обследованных нами пациентов ассоциации с клинической картиной заболевания не выявлено.
Данные, полученные с использованием панели генов, ассоциированных с развитием ЮМЭ, представлены в таблице 3.
Таблица 3. Патогенные и вероятно патогенные варианты, выявленные у пациентов с юношеской миоклонической эпилепсией (панель «ЮМЭ»)
Table 3. Pathogenic and likely pathogenic variants identified in patients with juvenile myoclonic epilepsy (JME panel)
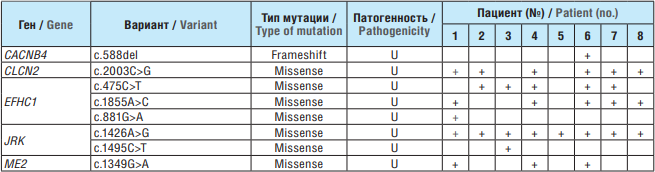
Примечание. U (англ. uncertain) – неясной патогенности.
Note. U – uncertain.
У обследованных нами пациентов выявлены миссенсмутации в генах CLCN2, EFHC1, JRK, ME2 и frameshiftмутация в гене CACNB4 (см. табл. 2). Патогенных вариантов в исследуемых генах, ассоциированных с развитием ЮМЭ, в ходе анализа обнаружено не было.
Большинство патогенных вариантов, ассоциированных с эпилепсией, располагаются в генах, кодирующих субъединицы нейрональных ионных каналов [30], что приводит к гипервозбудимости или недостаточности механизмов торможения нейронов головного мозга.
Ген GABRG2 кодирует гамма-субъединицу ГАМКрецепторов. В ряде публикаций сообщалось о следующих однонуклеотидных вариантах (ОНВ) гена GABRG2, ассоциированных с развитием различных форм ГГЭ: N79S (генетическая эпилепсия (ГЭ) с фебрильными приступами плюс), R82Q (ГЭ с фебрильными приступами плюс), P83S (ГЭ с фебрильными приступами плюс), R177G (GEFS+, ДАЭ), K328M (ГЭ с фебрильными приступами плюс) [31–34]. Эти варианты представляют собой миссенс-мутации, каждая из которых приводила к снижению поверхностной экспрессии субъединицы гамма-2 в комплексе ГАМК-рецептора типа А либо к изменению кинетических свойств канала. Кроме того, описаны нонсенс-мутации в гене GABRG2: Q40X (синдром Драве) [33][35][36], R136X (ГЭ с фебрильными приступами плюс) [37], Q390X (ГЭ с фебрильными приступами плюс, синдром Драве) [38], W429Х (ГЭ с фебрильными приступами плюс) [39]. Они приводят к снижению или полному отсутствию поверхностной экспрессии субъединицы гамма-2.
Помимо носительства ОНВ в некоторых исследованиях была обнаружена делеция c.1329delC гена GABRG2 (ГЭ с фебрильными приступами плюс) [40], появление которой вызывает экспрессию модифицированной субъединицы гамма-2 с удлиненным С-концевым доменом, образовавшимся в результате сдвига рамки считывания, последующего удаления естественного стоп-кодона и дальнейшей элонгации полипептидной цепочки путем транскрибирования некодирующей области. В результате происходит снижение гидрофобных свойств С-концевого домена субъединицы гамма-2.
Также описана мутация в сайте сплайсинга VS6+2T>G [41], приводящая к выпадению экзона 6 гена GABRG2 и преждевременному образованию стоп-кодона у пациентов с ДАЭ. ОНВ rs211037 (Asn196Asn) гена GABRG2 выявлен у пациентов с эпилепсией независимо от ее фенотипа – как с ЮМЭ, так и с мезиальной височной эпилепсией. Вариант rs211037 играет важную роль в регуляции транскрипции и регуляции сплайсинга данного гена [42].
Ген GABRA1 кодирует субъединицу альфа-1 ГАМКрецептора типа А. Среди миссенс-мутаций в данном гене обнаружены следующие патогенные варианты: exon9322 C>A, приводящий к замене аланина на аспарагин в позиции 322 у пациентов с диагностированной ЮМЭ [12], и 975delC, приводящий к преждевременной терминации трансляции в экзоне 8 гена GABRA1. Данный патогенный вариант ассоциирован с развитием ДАЭ [43][44]. Транслированный измененный белок имел пониженную стабильность из-за деградации, связанной с эндоплазматическим ретикулумом [43, 44]. ОНВ 659G>A гена GABRA1 снижает поверхностную экспрессию зрелого белка и/или эффективность нейротрансмиттера у пациентов с ЮМЭ и ДАЭ [45].
Инсерция K353delins18X гена GABRA1, представляющая собой вставку длиной 25 пар оснований, связана с сохранением интронной области в транскрипте, что также приводит к снижению поверхностной экспрессии ионного канала у пациентов с ЮМЭ и ДАЭ [45].
Ген EFHC1, расположенный на коротком плече 6-й хромосомы, кодирует белок, состоящий из 640 аминокислотных остатков и обладающий апоптозной активностью, с EF-hand мотивом, представляющим собой мотив связывания ионов кальция и не являющийся субъединицей ионного канала. У пациентов с ЮМЭ были обнаружены следующие миссенс-мутации в данном гене: 685T>C, 628G>A, 757G>T, 545G>A, 229C>A, 662G>A [46]. Эффект этих патогенных вариантов был изучен путем трансфекции клеточной культуры гиппокампа мыши вектором, содержащим дикий и мутантные варианты гена. В группе, трансфицированной мутантным вариантом, наблюдалось снижение клеточной смерти по сравнению с контрольной группой [46].
В семейных случаях ЮМЭ были выявлены ОНВ гена EFHC1: 685T>C, 1057C>T [47], 755C>A, 1523C>G; делеции: 789del.A, 362del.GAT; нонсенс-мутация: 829C>T [48]. Также в исследовании R. Thounaojam et al. (2017 г.) у пациентов с ЮМЭ обнаружены новые ОНВ 661C>T, 779G>A и 730C>T, приводящие к аминокислотным заменам и формированию преждевременного стоп-кодона: R221C, R260Q и R244STOP соответственно [49].
С другой стороны, D. Pinto et al. (2006 г.) сообщили об отсутствии патогенных ОНВ у 112 голландских пациентов с диагностированной ЮМЭ [50], что может свидетельствовать об отсутствии однозначной ассоциации гена EFHC1 с развитием ЮМЭ.
Ген CACNB4 кодирует субъединицу бета-4 потенциалзависимого кальциевого канала, играющего важнейшую роль в высвобождении нейротрансмиттеров в нейронах головного мозга. Субъединицы бета участвуют в трансмембранном переносе субъединиц альфа при сборке ионного канала. Ген CACNB4 располагается во 2-й хромосоме в локусе 2q22-23. Обнаружена нонсенс-мутация 1444C>T в гене CACNB4, приводящая к формированию преждевременного стоп-кодона R482X и элиминации 38 аминокислот С-концевого домена в семьях с ЮМЭ в качестве основного патологического фенотипа. В том же исследовании выявлена мутация 311G>T в гене CACNB4, приводящая к замене цистеина на фенилаланин C104F. Оба варианта вызывают увеличение скорости инактивации ионного канала [51].
CLCN2 – ген, кодирующий потенциал-зависимый хлоридный канал, располагается в локусе 3q26. Он сильно экспрессируется в головном мозге, особенно в нейронах, ингибируемых ГАМК. Предполагается, что CLCN2 играет важную роль в поддержании низкой внутриклеточной концентрации ионов хлора (Cl–), необходимой для ингибирующего ответа ГАМК. Мутации в данном локусе были найдены у пациентов с ЮМЭ, ДАЭ, ИГЭ с тонико-клоническими приступами пробуждения [52]. К. Haug et al. (2003 г.) обнаружили три типа гетерозиготных мутаций, приводящих к преждевременному формированию стопкодона, атипичному сплайсингу и небольшой аминокислотной замене [53]. Так, носительство варианта 597insG приводит к сдвигу рамки считывания и преждевременному формированию стоп-кодона в положении 231 [54]. Вариант IVS2-14del11 (делеция 11 пар оснований в интроне 2 рядом с сайтом сплайсинга) вызывает образование альтернативного продукта сплайсинга с утратой 44 аминокислотных остатков в экзоне 3. Миссенс-мутация 2144G>A обусловливает неконсервативную аминокислотную замену в С-концевом участке белка [54]. А миссенс-мутация 2154G>C в гене CLCN2 приводит к замене глутамина на аспарагин в позиции 718 [55].
Тем не менее, вопрос об ассоциации гена CLCN2 с ИГЭ, в т.ч. ЮМЭ, остается дискуссионным. Негативный эффект вышеописанных мутаций не воспроизвелся в исследовании M.I. Niemeyer (2009 г.) [56]. Кроме того, потенциально патогенные варианты гена CLCN2 были найдены и среди членов контрольных групп, поэтому мутации в CLCN2 могут быть лишь редкой причиной ИГЭ [57]. В исследовании H. Xie et al. (2019 г.) обнаружена новая миссенс-мутация 481G>A у пациента с ДАЭ, приводящая к замене глицина на серин и потенциально вызывающая изменение мотива селективного фильтра хлоридного ионного канала [58]. По-видимому, патогенные варианты гена CLCN2 являются минорными, их роль в патогенезе ИГЭ на сегодняшний день неясна.
Однако при популяционной стратификации ассоциированных с ЮМЭ вариантов, выявленных в исследованиях «случай–контроль», результаты не воспроизводились [19].
Ген BRD2 кодирует бромодоменсодержащий белок 2, локализованный на хромосоме 6p21.3. Нокаут данного гена у мышей приводит к нарушениям в формировании невральной трубки и головного мозга в эмбиогенезе, а гетерозиготное носительство мутаций в гене повышает риск эпилептогенеза [59]. Исследование ОНВ rs206787 и rs516535 гена BRD2 не выявило статистически значимых различий между больными ЮМЭ и контрольной группой [60][61].
Также в настоящее время возможными кандидатами, ассоциированными с ИГЭ, считаются ОНВ гена GABRB3: 1437T>G, 897T>C, 541T>C, 66 G>C, 75 G>A (ДАЭ) [62]; гена SLC12A5: L246P, G551D (ИГЭ с тонико-клоническими приступами пробуждения) [63]; гена SLC2A1: 696G>A, 641T>C (ДАЭ) [64].
К настоящему времени большинство изученных вариантов предрасположенности к ЮМЭ имеют недостаточную доказательную базу. Только rs2029461 гена GRM4, rs3743123 гена CX36 и rs3918149 гена BRD2 были связаны с ЮМЭ, по крайней мере, в двух независимых исследованиях генов-кандидатов [18]. А rs12059546 гена CHRM3 показал полногеномное значение для ЮМЭ. Однако эта положительная ассоциация не была воспроизведена в исследовании «случай–контроль», проведенном среди населения Китая [65].
В нашем исследовании не было идентифицировано ни одного патогенного или потенциально патогенного варианта в вышеуказанных генах.
Авторы выражают признательность компании Novel Software Systems (Россия) за оказанную помощь в анализе и интерпретации данных, полученных в ходе исследования.
В последние годы значительные усилия направлены на идентификацию генов предрасположенности к ЮМЭ. В ходе проведенного нами исследования моногенных и/или полигенных патогенных вариантов у обследованных пациентов с ЮМЭ и ребенка пробанда с ЮМЭ не выявлено. Высокая генетическая гетерогенность ЮМЭ может объяснить многочисленные безуспешные попытки найти гены предрасположенности к ЮМЭ. Необходимы дальнейшие исследования для подтверждения вариантов, ассоциированных с развитием ЮМЭ. Достижения в области геномных технологий могут расширить наше понимание генетики ЮМЭ.
1. Карлов В.А. Эпилепсия у детей и взрослых женщин и мужчин. Руководство для врачей. 2-е изд. М.: Бином; 2019: 896 с.
2. Marini C., Scheffer I. E., Crossland K.M., et al. Genetic architecture of idiopathic generalized epilepsy: clinical genetic analysis of 55 multiplex families. Epilepsia. 2004; 45 (5): 467–78. https://doi.org/10.1111/j.0013-9580.2004.46803.x.
3. Fisher R.S., Cross J.H., French J.A., et al. Operational classification of seizure types by the International League Against Epilepsy: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017; 58 (4): 522–30. https://doi.org/10.1111/epi.13670.
4. Hirsch E., French J., Scheffer I.E., et al. ILAE definition of the idiopathic generalized epilepsy syndromes: position statement by the ILAE Task Force on Nosology and Definitions. Epilepsia. 2022; 63 (6): 1475–99. https://doi.org/10.1111/epi.17236.
5. Scala M., Bianchi A., Bisulli F., et al. Advances in genetic testing and optimization of clinical management in children and adults with epilepsy. Expert Rev Neurother. 2020; 20 (3): 251–69. https://doi.org/10.1080/14737175.2020.1713101.
6. Helbig I. Genetic causes of generalized epilepsies. Semin Neurol. 2015; 35 (03): 288–92. https://doi.org/10.1055/s-0035-1552922.
7. Ноговицын В.Ю., Шарков А.А. ЭЭГ при генетических генерализованных эпилепсиях. Эпилепсия и пароксизмальные состояния. 2020; 12 (1S): S23–40. https://doi.org/10.17749/2077-8333.2020.12.1S.S23-S40.
8. Hempelmann A., Taylor K.P., Heils A., et al. Exploration of the genetic architecture of idiopathic generalized epilepsies. Epilepsia. 2006; 47 (10): 1682–90. https://doi.org/10.1111/j.1528-1167.2006.00677.x.
9. Vadlamudi L., Andermann E., Lombroso C.T., et al. Epilepsy in twins: insights from unique historical data of William Lennox. Neurology. 2004; 62 (7): 1127–33. https://doi.org/10.1212/01.wnl.0000118201.89498.48.
10. Corey L.A., Pellock J.M., Kjeldsen M.J., et al. Importance of genetic factors in the occurrence of epilepsy syndrome type: a twin study. Epilepsy Res. 2011; 97 (1-2): 103–11. https://doi.org/10.1016/j.eplepsyres.2011.07.018.
11. Wallace R.H., Marini C., Petrou S., et al. Mutant GABA(A) receptor gamma2-subunit in childhood absence epilepsy and febrile seizures. Nat Genet. 2001; 28 (1): 49–52. https://doi.org/10.1038/ng0501-49.
12. Cossette P., Liu L., Brisebois K., et al. Mutation of GABRA1 in an autosomal dominant form of juvenile myoclonic epilepsy. Nat Genet. 2002; 31 (2): 184–9. https://doi.org/10.1038/ng885.
13. Arsov T., Mullen S.A., Rogers S., et al. Glucose transporter 1 deficiency in the idiopathic generalized epilepsies. Ann Neurol. 2012; 72 (5): 807–15. https://doi.org/10.1002/ana.23702.
14. Scheffer I.E., Berkovic S., Capovilla G., et al. ILAE classification of the epilepsies: position paper of the ILAE Commission for Classification and Terminology. Epilepsia. 2017; 58 (4): 512–21. https://doi.org/10.1111/epi.13709.
15. Helbig I., Mefford H.C., Sharp A.J., et al. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009; 41 (2): 160–2. https://doi.org/10.1038/ng.292.
16. de Kovel C.G., Trucks H., Helbig I., et al. Recurrent microdeletions at 15q11.2 and 16p13.11 predispose to idiopathic generalized epilepsies. Brain. 2010; 133 (Pt. 1): 23–32. https://doi.org/10.1093/brain/awp262.
17. Dibbens L.M., Mullen S., Helbig I., et al. Familial and sporadic 15q13.3 microdeletions in idiopathic generalized epilepsy: precedent for disorders with complex inheritance. Hum Mol Genet. 2009; 18 (19): 3626–31. https://doi.org/10.1093/hmg/ddp311.
18. Santos B.P.D., Marinho C.R.M., Marques T.E.B.S., et al. Genetic susceptibility in juvenile myoclonic epilepsy: systematic review of genetic association studies. PLoS One. 2017; 12 (6): e0179629. https://doi.org/10.1371/journal.pone.0179629.
19. Mullen S.A., Berkovic S.F. ILAE Genetics Commission. Genetic generalized epilepsies. Epilepsia. 2018; 59 (6): 1148–53. https://doi.org/10.1111/epi.14042.
20. Шнайдер Н.А., Шилкина О.С., Петров К.В. и др. Клинико-генетическая гетерогенность юношеской миоклонической эпилепсии. Эпилепсия и пароксизмальные состояния. 2016; 8 (2): 20–36. https://doi.org/10.17749/2077-8333.2016.8.2.020-036.
21. Wang J., Lin Z.J., Liu L., et al. Epilepsy-associated genes. Seizure. 2017; 44: 11–20. https://doi.org/10.1016/j.seizure.2016.11.030.
22. National Library of Medicine. ClinVar. URL: https://www.ncbi.nlm.nih.gov/clinvar/ (дата обращения 23.04.2022).
23. Genome Aggregation Database (gnomAD). URL: https://gnomad.broadinstitute.org (дата обращения 23.04.2022).
24. Exome Aggregation Consortium (ExAC). URL: https://ngdc.cncb.ac.cn/databasecommons/database/id/3774 (дата обращения 23.04.2022).
25. OMIM®. An Online Catalog of Human Genes and Genetic Disorders. URL: https://www.omim.org (дата обращения 23.04.2022).
26. Ensembl. URL: https://www.ensembl.org/index.html (дата обращения 23.04.2022).
27. National Library of Medicine. National Center for Biotechnology Information. URL: https://www.ncbi.nlm.nih.gov (дата обращения 23.04.2022).
28. UniProt. URL: https://www.uniprot.org (дата обращения 23.04.2022).
29. Рыжкова О.П., Кардымон О.Л., Прохорчук Е.Б. и др. Руководство по интерпретации данных, полученных методами массового параллельного секвенирования (MPS). Медицинская генетика. 2017; 16 (7): 4–17.
30. Baulac S., Huberfeld G., Gourfinkel-An I., et al. First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet. 2001; 28 (1): 46–8. https://doi.org/10.1038/ng0501-46.
31. Huang X., Hernandez C.C., Hu N., et al. Three epilepsy-associated GABRG2 missense mutations at the γ+/β– interface disrupt GABAA receptor assembly and trafficking by similar mechanisms but to different extents. Neurobiol Dis. 2014; 68: 167–79. https://doi.org/10.1016/j.nbd.2014.04.015.
32. Shi X., Huang M.C., Ishii A., et al. Mutational analysis of GABRG2 in a Japanese cohort with childhood epilepsies. J Hum Genet. 2010; 55 (6): 375–8. https://doi.org/10.1038/jhg.2010.47.
33. Huang X., Tian M., Hernandez C.C., et al. The GABRG2 nonsense mutation, Q40X, associated with Dravet syndrome activated NMD and generated a truncated subunit that was partially rescued by aminoglycoside-induced stop codon read-through. Neurobiol Dis. 2012; 48 (1): 115–23. https://doi.org/10.1016/j.nbd.2012.06.013.
34. Ishii A., Kanaumi T., Sohda M., et al. Association of nonsense mutation in GABRG2 with abnormal trafficking of GABAA receptors in severe epilepsy. Epilepsy Res. 2014; 108 (3): 420–32. https://doi.org/10.1016/j.eplepsyres.2013.12.005.
35. Ishii A., Kanaumi T., Sohda M., et al. Association of nonsense mutation in GABRG2 with abnormal trafficking of GABAA receptors in severe epilepsy. Epilepsy Res. 2014; 108 (3): 420–32. https://doi.org/10.1016/j.eplepsyres.2013.12.005.
36. Hirose S. A new paradigm of channelopathy in epilepsy syndromes: intracellular trafficking abnormality of channel molecules. Epilepsy Res. 2006; 70 (1): S206–17. https://doi.org/10.1016/j.eplepsyres.2005.12.007.
37. Johnston A.J., Kang J.Q., Shen W., et al. A novel GABRG2 mutation, p.R136*, in a family with GEFS+ and extended phenotypes. Neurobiol Dis. 2014; 64: 131–41. https://doi.org/10.1016/j.nbd.2013.12.013.
38. Harkin L.A., Bowser D.N., Dibbens L.M., et al. Truncation of the GABA(A)-receptor gamma2 subunit in a family with generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2002; 70 (2): 530–6. https://doi.org/10.1086/338710.
39. Sun H., Zhang Y., Liang J., et al. SCN1A, SCN1B, and GABRG2 gene mutation analysis in Chinese families with generalized epilepsy with febrile seizures plus. J Hum Genet. 2008; 53 (8): 769–74. https://doi.org/10.1007/s10038-008-0306-y.
40. Tian M., Mei D., Freri E., et al. Impaired surface αβγ GABA(A) receptor expression in familial epilepsy due to a GABRG2 frameshift mutation. Neurobiol Dis. 2013; 50: 135–41. https://doi.org/10.1016/j.nbd.2012.10.008.
41. Kananura C., Haug K., Sander T., et al. A splice-site mutation in GABRG2 associated with childhood absence epilepsy and febrile convulsions. Arch Neurol. 2002; 59 (7): 1137–41. https://doi.org/10.1001/archneur.59.7.1137.
42. Balan S., Sathyan S., Radha SK., et al. GABRG2, rs211037 is associated with epilepsy susceptibility, but not with antiepileptic drug resistance and febrile seizures. Pharmacogenet Genomics. 2013; 23 (11): 605–10. https://doi.org/10.1097/FPC.0000000000000000.
43. Kang J.Q., Shen W., Macdonald R.L. Two molecular pathways (NMD and ERAD) contribute to a genetic epilepsy associated with the GABAA receptor GABRA1 PTC Mutation, 975delC, S326fs328X. J Neurosci. 2009.; 29 (9): 2833–44. https://doi.org/10.1523/jneurosci.4512-08.2009.
44. Maljevic S., Krampfl K., Cobilanschi J., et al. A mutation in the GABA(A) receptor alpha(1)-subunit is associated with absence epilepsy. Ann Neurol. 2006; 59 (6): 983–7. https://doi.org/10.1002/ana.20874.
45. Lachance-Touchette P., Brown P., Meloche C., et al. Novel α1 and γ2 GABAA receptor subunit mutations in families with idiopathic generalized epilepsy. Eur J Neurosci. 2011; 34 (2): 237–49. https://doi.org/10.1111/j.1460-9568.2011.07767.x.
46. Suzuki T., Delgado-Escueta A.V., Aguan K., et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet. 2004; 36 (8): 842–9. https://doi.org/10.1038/ng1393.
47. Annesi F., Gambardella A., Michelucci R., et al. Mutational analysis of EFHC1 gene in Italian families with juvenile myoclonic epilepsy. Epilepsia. 2007; 48 (9): 1686–90. https://doi.org/10.1111/j.1528-1167.2007.01173.x.
48. Medina M.T., Suzuki T., Alonso M.E., et al. Novel mutations in Myoclonin1/EFHC1 in sporadic and familial juvenile myoclonic epilepsy. Neurology. 2008; 70 (22 Pt. 2): 2137–44. https://doi.org/10.1212/01.wnl.0000313149.73035.99.
49. Thounaojam R., Langbang L., Itisham K., et al. EFHC1 mutation in Indian juvenile myoclonic epilepsy patient. Epilepsia Open. 2017; 2 (1): 84–9. https://doi.org/10.1002/epi4.12037.
50. Pinto D., Louwaars S., Westland B., et al. Heterogeneity at the JME 6p11-12 locus: absence of mutations in the EFHC1 gene in linked Dutch families. Epilepsia. 2006; 47 (10): 1743–6. https://doi.org/10.1111/j.1528-1167.2006.00676.x.
51. Escayg A., De Waard M., Lee D.D., et al. Coding and noncoding variation of the human calcium-channel β4-subunit gene CACNB4 in patients with idiopathic generalized epilepsy and episodic ataxia. Am J Hum Genet. 2000; 66 (5): 1531–9. https://doi.org/10.1086/302909.
52. D’Agostino D., Bertelli M., Gallo S., et al. Mutations and polymorphisms of the CLCN2 gene in idiopathic epilepsy. Neurology. 2004; 63 (8): 1500–2. https://doi.org/10.1212/01.wnl.0000142093.949.
53. Haug K., Warnstedt M., Alekov A.K., et al. Mutations in CLCN2 encoding a voltage-gated chloride channel are associated with idiopathic generalized epilepsies. Nat Genet. 2003; 33 (4): 527–32. https://doi.org/10.1038/ng1121.
54. Kleefuß-Lie A., Friedl W., Cichon S., et al. CLCN2 variants in idiopathic generalized epilepsy. Nat Genet. 2009; 41 (9): 954–5. https://doi.org/10.1038/ng0909-954.
55. Everett K., Chioza B., Aicardi J., et al. Linkage and mutational analysis of CLCN2 in childhood absence epilepsy. Epilepsy Res. 2007; 75 (2-3): 145–53. https://doi.org/10.1016/j.eplepsyres.2007.05.004.
56. Niemeyer M.I., Cid L.P., Sepúlveda F.V., et al. No evidence for a role of CLCN2 variants in idiopathic generalized epilepsy. Nat Genet. 2010; 42 (1): 3. https://doi.org/10.1038/ng0110-3.
57. Stogmann E., Lichtner P., Baumgartner C., et al. Mutations in the CLCN2 gene are a rare cause of idiopathic generalized epilepsy syndromes. Neurogenetics. 2007; 7 (4): 265–8. https://doi.org/10.1007/s10048-006-0057-x.
58. Xie H., Su W., Pei J., et al. De novo SCN1A, SCN8A, and CLCN2 mutations in childhood absence epilepsy. Epilepsy Res. 2019; 154: 55–61. https://doi.org/10.1016/j.eplepsyres.2019.04.
59. Shang E., Wang X., Wen D., et al. Double bromodomain-containing gene Brd2 is essential for embryonic development in mouse. Dev Dyn. 2009; 238 (4): 908–17. https://doi.org/10.1002/dvdy.21911.
60. Shilkina О.S., Zobova S.N., Domoratskaya Е.А., Dmitrenko D.V. Clinical and genetic characteristics of juvenile myoclonic epilepsy. Personalized Psychiatry and Neurology. 2021; 1 (2): 95–105. https://doi.org/10.52667/2712-9179-2021-1-2-95-105.
61. Шилкина О.С., Шнайдер Н.А., Зобова С.Н. и др. Ассоциация носительства полиморфизмов rs206787 и rs516535 гена BRD2 и rs3743123 гена GJD2 с юношеской миоклонической эпилепсией у пациентов европейского происхождения в Сибири. Неврология, нейропсихиатрия, психосоматика. 2019; 11 (4): 61–7. https://doi.org/10.14412/2074-2711-2019-4-61-67.
62. Tanaka M., Olsen R.W., Medina M.T., et al. Hyperglycosylation and reduced GABA currents of mutated GABRB3 polypeptide in remitting childhood absence epilepsy. Am J Hum Genet. 2008; 82 (6): 1249–61. https://doi.org/10.1016/j.ajhg.2008.04.020.
63. Kearney J.A. Locus heterogeneity in epilepsy of infancy with migrating focal seizures. Epilepsy Curr. 2016; 16 (1): 43–5. https://doi.org/10.5698/1535-7597-16.1.43.
64. Larsen J., Johannesen K.M., Ek J., et al. The role of SLC2A1 mutations in myoclonic astatic epilepsy and absence epilepsy, and the estimated frequency of GLUT1 deficiency syndrome. Epilepsia. 2015; 56 (12): e203–8. https://doi.org/10.1111/epi.13222.
65. Zhang Y., Qu J., Mao C.X., et al. Novel susceptibility loci were found in Chinese genetic generalized epileptic patients by genome-wide association study. CNS Neurosci Ther. 2014; 20 (11): 1008–10. https://doi.org/10.1111/cns.12328.
Тимечко Елена Евгеньевна – лаборант лаборатории медицинской генетики центра коллективного пользования «Молекулярные и клеточные технологии»
WoS ResearcherID: CAF-2677-2022; РИНЦ SPIN-код: 2711-7770
ул. Партизана Железняка, д. 1, Красноярск 660022
Шилкина Ольга Сергеевна – к.м.н., невролог Неврологического центра эпилептологии, нейрогенетики и исследований мозга Университетской клиники
РИНЦ SPIN-код: 1150-7413
ул. Партизана Железняка, д. 1, Красноярск 660022
Орешкова Наталья Викторовна – к.б.н., заведующая лабораторией геномных исследований и биотехнологии; старший научный сотрудник лаборатории лесной геномики Научно-образовательного центра геномных исследований Института фундаментальной биологии и биотехнологии
WoS ResearcherID: L-5516-2017; Scopus Author ID: 55793767200; РИНЦ SPIN-код: 4149-9633
ул. Академгородок, д. 50, Красноярск 660036; пр. Свободный, д. 79, Красноярск 660041
Кобаненко Владислав Олегович – студент 5-го курса (медицинская кибернетика)
РИНЦ SPIN-код: 1143-4417
ул. Партизана Железняка, д. 1, Красноярск 660022
Осипова Елизавета Алексеевна – студентка 5-го курса (медицинская кибернетика)
ул. Партизана Железняка, д. 1, Красноярск 660022
Шнайдер Наталья Алексеевна – д.м.н., профессор, ведущий научный сотрудник центра коллективного пользования «Молекулярные и клеточные технологии» ; ведущий научный сотрудник отделения персонализированной психиатрии и неврологии
WoS ResearcherID: M-7084-2014; РИНЦ SPIN-код: 1952-3043
ул. Партизана Железняка, д. 1, Красноярск 660022; ул. Бехтерева, д. 3, Санкт-Петербург 192019
Дмитренко Диана Викторовна – д.м.н., заведующая кафедрой медицинской генетики и клинической нейрофизиологии
Института профессионального образования
WoS ResearcherID: H-7787-2016; РИНЦ SPIN-код: 9180-6623
ул. Партизана Железняка, д. 1, Красноярск 660022
Тимечко Е.Е., Шилкина О.С., Орешкова Н.В., Кобаненко В.О., Осипова Е.А., Шнайдер Н.А., Дмитренко Д.В. Полноэкзомное секвенирование пациентов с юношеской миоклонической эпилепсией. Эпилепсия и пароксизмальные состояния. 2022;14(3):254-266. https://doi.org/10.17749/2077-8333/epi.par.con.2022.119
Timechko E.E., Shilkina O.S., Oreshkova N.V., Kobanenko V.O., Osipova E.A., Shnayder N.A., Dmitrenko D.V. Whole-exome sequencing of patients with juvenile myoclonic epilepsy. Epilepsy and paroxysmal conditions. 2022;14(3):254-266. (In Russ.) https://doi.org/10.17749/2077-8333/epi.par.con.2022.119

101000 г. Москва, вн.тер.г. муниципальный округ Басманный, пер. Лялин, д. 11-13/1, стр. 3
Тел.: +7(495)6495495
e-mail: info@irbis-1.ru



































