


ОРИГИНАЛЬНЫЕ СТАТЬИ
Актуальность. В ситуациях, когда консервативная противоэпилептическая терапия неэффективна, возможно хирургическое лечение, направленное на удаление эпилептогенного очага. Резекционные операции позволяют избавиться от приступов в большинстве случаев, однако у 20–30% больных они сохраняются или рецидивируют. В таких случаях пациентам может быть предложен тот или иной вид нейромодуляции.
Цель: оценка эффективности проведения нейромодуляции у пациентов с фармакорезистентной эпилепсией (ФРЭ) после неудачных резекционных хирургических вмешательств.
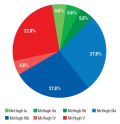
Материал и методы. Проведен ретроспективный анализ данных 23 пациентов, которым после неудачной операции по поводу ФРЭ проводилась стимуляция блуждающего нерва (англ. vagus nerve stimulation, VNS) либо глубокая стимуляция (англ. deep brain stimulation, DBS) переднего ядра таламуса (англ. anterior nucleus of the thalamus, ANT) или гиппокампа (англ. hippocampus, HP). Система VNS имплантирована 18 (78,3%) больным, система HP-DBS – 3 (13,0%), система ANT-DBS – 2 (8,7%). Результаты хирургических вмешательств оценивали по шкале Engel, VNS-терапии – по шкале McHugh (MH), DBS-терапии – по степени снижения частоты приступов в процентах. Средний катамнез наблюдения составил 56,5 мес.
Результаты. В группе пациентов, которым была имплантирована система VNS, у 3 (16,7%) исход составил MH Ia–IIb, у 10 (55,5%) – MH IIIa–IIIb, у 5 (27,8%) – MH IV–V. В группе HP-DBS у 2 больных из 3 отмечено снижение частоты приступов более чем на 50% от исходного уровня, у 1 пациента наблюдалось облегчение тяжести приступов. В группе ANT-DBS у одного больного зарегистрировано снижение частоты приступов на 60% и облегчение тяжести приступов, у второго изменений в частоте приступов не отмечено.
Заключение. Нейромодуляция у пациентов с ФРЭ позволяет значимо уменьшить частоту приступов у более чем половины пациентов после неудачного оперативного лечения.

Цель: изучить влияние вальпроата натрия на концентрации гомоцистеина, фолата и витамина B12 в плазме крови у больных с длительно присутствующей эпилепсией с тонико-клоническими приступами в сравнении с пациентами с впервые диагностированной эпилепсией и контрольной группой.
Материал и методы. В исследование включены 90 участников (средний возраст 36,30±12,83 года, большинство (58,89%) составляли мужчины), которые были разделены на три группы: 30 человек, не страдающих эпилепсией (контрольная 1-я группа), 30 больных с впервые выявленной эпилепсией (2-я группа) и 30 пациентов с длительно присутствующей эпилепсией с тонико-клоническими приступами (3-я группа). В 3-й группе больные получали вальпроат натрия. Все участники исследования прошли клиническое и неврологическое обследование. Различия в уровнях гомоцистеина, фолиевой кислоты, витамина B12 в плазме крови в трех группах исследовали через 6 мес наблюдения.
Результаты. Зарегистрирован повышенный уровень гомоцистеина во 2-й и 3-й группах; во 2-й группе он был достоверно выше, чем в 3-й и 1-й группах (р=0,001). Уровень фолатов в плазме во 2-й и 3-й группах оказался снижен; в 3-й группе он был достоверно выше, чем во 2-й группе, и ниже, чем в 1-й группе (р=0,001). Снижение уровня витамина В12 во 2-й и 3-й группах было недостоверно (р=0,090). В 1-й и 2-й группах наблюдалась значимая корреляция между показателями.
Заключение. Введение вальпроата натрия может нарушить гомеостатические уровни гомоцистеина, фолата и витамина В12 и вызвать их колебания в сыворотке крови у больных с длительно присутствующей эпилепсией с тонико-клоническими приступами.
Актуальность. Соли лития используются в психиатрии и, в зависимости от аниона, могут проявлять нейропротекторные эффекты.
Цель: изучить влияние аскорбата лития (LiAsc) и карбоната лития в различных дозах per os на выраженность и тяжесть течения у крыс первично-генерализованных судорог, вызванных тиосемикарбазидом in vivo.
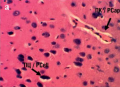
Материал и методы. Исследование проведено на 30 белых крысах-самцах линии Вистар массой 200–300 г в пяти группах: контроль, карбонат лития в дозе 5 мг/кг, LiAsc в дозах 5, 10, 15 мг/кг/сут, вводившиеся зондированием per os в течение 14 сут. Модель первично-генерализованных судорог воспроизведена введением 28 мг/кг тиосемикарбазида внутрибрюшинно. Степень неврологического дефицита оценивали по показателям судорог (латентный период до судорог, количество судорожных приступов и вздрагиваний, клонические судороги и др.). Проведены патогистологические исследования ткани головного мозга с морфометрией гистологических срезов на анализаторе изображения BioVision (Австрия).
Результаты. Курсовое введение и карбоната, и LiAsc статистически значимо снижало длительность судорог. Использование LiAsc в дозах 10–15 мг/кг/сут достоверно не только сокращало длительность судорог (р=0,01), но и увеличивало латентный период до них (р=0,03), уменьшало число приступов (р<0,05) и частоту встречаемости тонической экстензии (р=0,01).
Заключение. Доза LiAsc 10 мг/кг/сут достаточна для снижения неврологического дефицита при воспроизведении модели тиосемикарбазидных судорог, что подтверждается результатами патогистологического исследования и морфометрического анализа образцов головного мозга.
КЛИНИЧЕСКИЕ СЛУЧАИ
Синдром нарушения развития нервной системы PACS1 (синдром Схюрс-Хоймакерса (англ. Schuurs-Hoeijmakers syndrome); MIM 615009) – редкое аутосомно-доминантное генетическое заболевание, характеризующееся задержкой развития, интеллектуальным дефицитом, дисморфическими чертами, а иногда и судорогами. В статье представлен клинический случай синдрома PACS1 у пациентки с задержкой психоречевого и моторного развития, эпилепсией и описанными вариантами в гене PACS1 (rs398123009, chr11:6621120, c.607C>T, p.Arg203Trp). Знание молекулярных механизмов развития синдрома PACS1 важно не только для генотип-фенотипической корреляции, но и для разработки новых терапевтических подходов в лечении, которые могли бы улучшить качество жизни пациентов.
Представлен клинический пример пациента с синдромом Драве, вызванным мутацией в гене SCN1A. Синдром Драве – это тяжелая эпилептическая энцефалопатия, встречающаяся в раннем детском возрасте и сопровождающаяся полиморфизмом приступов, фармакорезистентным течением эпилепсии, грубыми когнитивными нарушениями. Данный случай подтверждает возможность медикаментозного контроля течения синдрома Драве. Достигнута 2-летняя ремиссия на фоне применения политерапии антиэпилептическими препаратами. В настоящее время сохраняется ремиссия на дуотерапии: топирамат в сочетании с леветирацетамом. Описанный клинический случай также демонстрирует сохранность когнитивных функций: ребенок успешно, в полном объеме осваивает общеобразовательную программу. Следует отметить, что при раннем купировании эпилептических приступов когнитивные функции не страдают.
Транзиторная глобальная амнезия (ТГА) и транзиторная эпилептическая амнезия (ТЭА) – редко встречающиеся в клинической практике феномены, которые проявляются преходящими когнитивными амнестическими нарушениями. Несмотря на схожесть клинической картины, эти состояния патогенетически разнородны и требуют различных терапевтических подходов. ТГА – клинический синдром, характеризующийся внезапной антероградной амнезией события длительностью до 24 ч без очаговых неврологических симптомов, не склонный к рецидивированию. Под маской ТГА часто протекает TЭA – эпилептические приступы с нарушением осознанности различной длительности, включая долгосрочные (более 24 ч), как вариант фокальной эпилепсии. Для ТЭА характерны повторяемость эпизодов, сочетание с другими проявлениями эпилепсии, коморбидность с нейродегенеративными заболеваниями (деменцией). Для дифференциальной диагностики необходимо использовать пролонгированный видеоэлектроэнцефалографический мониторинг с записью сна, методы нейровизуализации (магнитно-резонансную томографию, позитронно-эмиссионную томографию головного мозга), психологическое тестирование, биохимическое исследование маркеров нейродегенерации.
НАУЧНЫЕ ОБЗОРЫ

Болезнь де Виво является редким генетическим нарушением, связанным с дефицитом транспортера глюкозы 1-го типа (англ. glucose transporter type 1, GLUT1). В статье представлен обзор публикаций, в которых описаны различные клинические проявления заболевания, включая сочетание эпилепсии с хореическим гиперкинезом. Отмечается резистентность приступов к базовой антиэпилептической терапии, в качестве основного метода лечения предлагается кетогенная диета. Приведено собственное клиническое наблюдение 18-летнего пациента, у которого с 1,5 лет впервые возникли миоклонико-астатические приступы и атактические проявления в виде нарушения координации движений и неустойчивости при ходьбе. На фоне терапии препаратами вальпроевой кислоты приступы сохранялись с частотой до 5 раз в месяц. С 17 лет появились непроизвольные насильственные нерегулярные движения мышц туловища и конечностей, совершающиеся в быстром темпе. Проведено комплексное обследование, в результате верифицирован диагноз «болезнь де Виво». Путем назначения адекватной противоэпилептической терапии и кетогенной диеты удалось стабилизировать состояние больного, купировать проявления эпилепсии и гиперкинетического расстройства. Мы обращаем внимание специалистов на дифференциальную диагностику состояний, характеризующихся эпилептическими приступами, умственной отсталостью и насильственными движениями, а также на диагностику и тактику ведения пациентов с болезнью де Виво. К сожалению, не все больные с данной патологией получают адекватную патогенетическую и симптоматическую терапию. Зачастую пациенты подвергаются многочисленным госпитализациям, т.к. не диагностирована основная причина симптомов, а именно дефицит GLUT1.

Эпилепсия относится к одним из наиболее древних заболеваний. Пациенты, страдающие эпилепсией, с античных времен сталкивались с проблемой стигматизации и дискриминации, т.к. медицинские знания в то время были ограниченны и болезнь связывали с различными мистическими и мифическими явлениями, а лечение отсутствовало. Статья широко освещает вопросы стигматизации эпилепсии, охватывая весь продолжительный исторический путь заболевания и обозначая явление стигматизации как серьезную социально значимую проблему. Многие больные эпилепсией страдают не только из-за симптомов болезни, но и из-за общественной дискриминации, что значительно снижает их качество жизни и приводит к социальной дезадаптации. К сожалению, в настоящее время проблема стигматизации эпилепсии полностью не исчезла и осталась не только в развивающихся, но и развитых странах. Цель данной работы заключалась в демонстрации актуальности этого вопроса и повышении осведомленности о нем. Времена, когда пациенты с эпилепсией считались одержимыми духами, проклятыми и неизлечимыми, остались далеко позади, и в современном обществе любая форма социальной дискриминации таких людей недопустима.

Контент доступен под лицензией Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License.
ISSN 2311-4088 (Online)



































